INTRODUÇÃO
A aterosclerose é um processo crônico predominantemente silencioso em sua evolução, no qual fatores locais e sistêmicos interagem de forma a abreviar ou retardar eventos clínicos. Na doença obstrutiva de carótidas, o caráter catastrófico do acidente vascular encefálico (AVE), atualmente uma prioridade em saúde pública no mundo desenvolvido, justifica a emergente preocupação com características morfológicas, inflamatórias e bioquímicas da placa ateromatosa, que eventualmente possam conferir vulnerabilidade às lesões.
Atualmente, sabe-se que uma agressão sistemática promove respostas compensatórias que alteram a homeostase do endotélio, sobretudo por meio da ativação de leucócitos e plaquetas, e da alteração de permeabilidade. As células endoteliais secretam moléculas vasoativas de adesão, citoquinas e fatores de crescimento, entre outras substâncias, as quais, se o processo de agressão não é bloqueado, continuam a serem produzidas indefinidamente. Igualmente, a resposta inflamatória induz à migração e à proliferação de células musculares lisas para a porção subintimal, por meio de mediadores químicos liberados por macrófagos modificados pela captação de lipídeos (células espumosas), e por subtipos específicos de linfócitos T. Nesta fase, o crescimento intraparietal da área de inflamação ocorre no sentido oposto à luz da artéria, em direção à adventícia, sempre estimulando a liberação de enzimas proteolíticas da matriz intersticial, de citoquinas, e de fator de crescimento tumoral, o que eventualmente pode induzir necrose focal [1,2]. Além disso, o processo de acúmulo dos macrófagos está relacionado ao aumento da concentração plasmática de interleucinas, proteína C reativa, e de outros marcadores inflamatórios, propostos atualmente como sinalizadores da presença ou da instabilidade clínica na aterosclerose, tanto em nível local quanto sistêmico [3].
Também o possível papel de agentes infecciosos, como a clamídia, tem sido investigado na gênese das placas de ateroma, ou no desencadeamento de eventos, com dados ainda inconclusivos [4-6].
Outra manifestação da atividade imuno-inflamatória na doença aterosclerótica diz respeito à produção de auto-anticorpos que agridem células endoteliais e/ou células musculares lisas da parede arterial. Estudos experimentais e clínicos têm proposto que o aumento dos níveis séricos de anticorpos contra fosfolipídeos da membrana celular, ou contra proteínas de choque térmico, esteja associado a eventos agudos na aterosclerose, embora a relação causal ainda não esteja bem elucidada [7-10].
Além disso, o aprimoramento dos métodos de imagem não invasivos, como a ultra-sonografia e a angiografia por ressonância nuclear magnética, tem proporcionado maior conhecimento da estrutura morfológica da placa de carótida, e sua possível associação com desfechos clínicos. A capacidade destes métodos em identificar sinais de instabilidade, a partir da análise do conteúdo e da superfície da placa, tem sido recentemente testada em diversos estudos de correspondência histológica [11].
A parte 1 desta revisão analisou estudos recentes, relacionados à vulnerabilidade na doença aterosclerótica de artérias carótidas, expressa em seus aspectos epidemiológicos, clínicos e inflamatórios. Nesta parte, serão abordados os marcadores séricos de uso corrente, bem como aspectos histológicos, e de diagnóstico por imagem, relativos à instabilidade da doença carotídea, discutindo possíveis implicações terapêuticas ou eventuais modificações de paradigmas relacionados às indicações atuais de intervenção.
Marcadores sorológicos
Nos últimos anos, particular interesse tem ocorrido na identificação de mediadores inflamatórios circulantes, associados aos processos ateroscleróticos e à predição de eventos. Possíveis implicações terapêuticas, como o papel das estatinas, do ácido acetilsalicílico e dos inibidores da cicloxigenase, têm sido igualmente estudadas [12,13].
Na doença obstrutiva de carótida, trabalhos recentes têm procurado esclarecer o papel das várias substâncias envolvidas na formação, no crescimento e possivelmente na instabilização da placa. Embora Waddington et al. [14], estudando a oxidação de ácidos graxos na doença carotídea, não tenham conseguido demonstrar sua correlação direta com a instabilidade clínica, Nishi et al. [15] observaram que níveis elevados de anticorpos anti-LDL oxidado tiveram correspondência com maior infiltração de macrófagos na análise histológica de 44 espécimes de EC, sugerindo que este marcador possa conferir vulnerabilidade à placa. Incontroverso, entretanto, parece ser o papel protetor do HDL-colesterol na progressão da doença carotídea. Johnsen et al. [16], acompanhando por até sete anos 1952 pacientes com doença carotídea diagnosticada por ultra-sonografia, evidenciaram taxa menor de crescimento das lesões no grupo no quartil superior dos níveis de HDL. Este achado vem ao encontro do observado tanto em relação à recorrência de desfechos coronários [17], como cerebrais [18].
Outro sinalizador de interesse na resposta imunológica que acompanha o processo aterosclerótico é o complexo CD40 e CD40 ligante. Inicialmente relacionados à atividade exclusivamente linfocitária, estes marcadores já têm sua expressão documentada em outras células presentes na formação da placa de ateroma. Há evidências de que a interação CD40 e CD40L contribui para instabilização da placa, por meio da interação com células endoteliais, células musculares lisas, macrófagos e plaquetas, promovendo a liberação de citoquinas, fator de necrose tumoral alfa, metaloproteinases de matriz extracelular e substâncias pró-coagulantes [19]. Uma subanálise do Women's Health Study demonstrou níveis significativamente elevados de CD40L em 130 mulheres que desenvolveram infarto agudo do miocárdio ou AVE no período de quatro anos, em comparação a participantes fumantes, pareadas para a idade, mas com níveis de CD40L normais (p = 0,02) [20]. Em outro estudo, a análise de 2908 casos de síndromes coronarianas agudas demonstrou maior risco de recorrência de eventos, no período de 16 semanas, quando os níveis de CD40L encontravam-se acima do percentil 90 [21]. A hipótese de que níveis plasmáticos de CD40L relacionam-se com características morfológicas de vulnerabilidade foi testada por Blake et al. [22], em 49 pacientes com estenose de carótida. Embora os níveis de CD40L não tenham se correlacionado ao porcentual de estenose, CD40L acima da média se associou com a presença de lagos lipídicos acelulares intraplaca.
Estudos de correspondência histológica também têm se proposto ao estudo da eventual ação das metaloproteinases de matriz extracelular na ateromatose de carótida. Sapienza et al. [23], analisando 53 casos consecutivos de EC, demonstraram que nos pacientes com lesões classificadas microscopicamente como instáveis, os níveis séricos de diversas metaloproteinases foram significativamente maiores do que nas lesões estáveis. Entretanto, por meio do doppler transcraniano, Loftus et al. [24] observaram que níveis elevados da metaloproteinase de matriz tipo 9 relacionaram-se à maior ocorrência de microembolismo intra-operatório, durante EC, enquanto Molloy et al. [25] conseguiram demonstrar a correlação entre elevação de metaloproteinase de matriz tipo 8, sintomas recentes, embolização silenciosa e evidências histológicas de ruptura da placa. Recentemente, Sluijter et al. [26] propuseram que a atividade das diferentes MMPs pode variar de acordo com o predomínio celular da placa de ateroma. A análise de 150 espécimes de EC revelou atividade significativamente maior das metaloproteinase de matriz tipo 8 e 9 nas lesões ricas em macrófagos, e da metaloproteinase de matriz tipo 2 em placas com predomínio de células musculares lisas. A correlação entre os elementos celulares, sintomas e níveis de metaloproteinase de matriz tipo 8 e 9 também foi testada por Verhoeven et al. [27], em 404 pacientes submetidos à cirurgia de carótida. Nos casos com AIT ou AVE recentes, os níveis de metaloproteinases foram significativamente maiores do que os observados nos pacientes assintomáticos (p<0,001). No entanto, embora pareça haver relação direta entre a elevação destes marcadores e vulnerabilidade da placa, não há ainda evidências para seu emprego como preditor de risco na prática clínica.
Outro grupo de substâncias com atividade comprovada na doença ateromatosa, tanto em nível tecidual quanto plasmático, são as interleucinas [28]. Embora a maioria das interleucinas tenha papel pró-inflamatório, sendo as mais estudadas as interleucinas-1,-2,-6,-7,-8 e -18, alguns subgrupos parecem ter qualidades antiinflamatórias, o que determina uma modulação do efeito resultante e produz oscilações durante o curso da doença. Atividade marcadamente mais elevada da interleucina-18 foi observada por Mallat et al. [29] em placas de carótida sintomáticas ou ulceradas, em comparação a casos considerados estáveis. Também foi demonstrado seu efeito estimulante na produção de fator de necrose tumoral alfa, de interferon e moléculas de adesão, ou indutor da apoptose celular, a partir da interação com a interleucina-12 [30]. Recentemente, a relação entre elevados níveis de interleucina-18 circulante e adelgaçamento da capa fibrosa em placas de carótida foi demonstrada por Yamagami et al. [31], em 366 pacientes sem eventos cerebrovasculares prévios, achados comparáveis aos descritos por Elkind et al. [32], em relação à interleucina-2. Embora com menor consistência, o possível efeito antiinflamatório de alguns tipos de interleucinas também tem sido investigado. Em análise histoquímica preliminar de 21 espécimes de EC, demonstrou-se relação inversa entre a expressão da interleucina-10 com a produção de óxido nítrico sintetase e a taxa de apoptose celular, sugerindo um efeito protetor contra a excessiva necrose intraplaca e, supostamente, conferindo estabilidade às lesões [33].
Dentre todas as substâncias inflamatórias associadas à aterogênese, a proteína C reativa (PRC) tem se afirmado como o mais importante biomarcador de eventos cardiovasculares, principalmente após a descoberta da forma de aferição ultra-sensível (PCR-us) [34,35]. A PCR é um polipeptídeo secretado pelo fígado, principalmente, em resposta à elevação dos níveis plasmáticos de interleucina-6, que apresenta ação comprovada na diferenciação dos macrófagos em células espumosas [36], na produção de moléculas de adesão e selectinas [37], no recrutamento local de monócitos [38], na ativação do complemento [39], na destruição celular mediada por linfócitos T [40], na potencialização dos efeitos da angiotensina II [40] e na atenuação da produção e da ação do óxido nítrico [41].
A associação entre níveis elevados de PCR-us e prognóstico cardiovascular foi primeiramente demonstrada nas síndromes coronarianas agudas (SCA), a partir da evidência de que níveis de PCR-us ? 0.3g/dl relacionaram-se a mais episódios isquêmicos, e pior evolução, tanto precoce quanto tardiamente [42,43]. Em pacientes após infarto agudo do miocárdio, o estudo CARE [44] revelou risco aumentado de eventos recorrentes ou morte, na presença de níveis elevados de PCR-us, resultado consonante com o de outros autores [45,46].
Quando associado ao escore de Framingham e aos níveis de LDL colesterol, a PCR-us pode adquirir valor preditivo adicional, na evolução da doença coronariana [47,48]. Além disso, a PCR-us tem sido apontada em vários estudos como preditor de morte súbita, infarto cerebral ou desenvolvimento de doença arterial obstrutiva de membros inferiores, ainda que com um efeito menos robusto [49-51].
O papel da PCR-us na predição de eventos cerebrovasculares foi estudado, em uma coorte do estudo de Framingham, que correlacionou a incidência do primeiro episódio de AIT ou AVE, com quartis séricos de PCR-us, ao longo de 14 anos. Nos 1462 casos avaliados, o quartil superior duplicou o risco relativo dos desfechos nos homens, e triplicou nas mulheres, independente da idade, embora na amostra predominassem indivíduos idosos (média 70 anos) [50]. Também tem sido sugerido que a PCR-us elevada pode ser preditor independente de morte e eventos recorrentes, após o primeiro episódio de AVE [52,53].
Sem analisar desfechos clínicos, Wang et al. [54] demonstraram que o desenvolvimento de espessamento médio-intimal da carótida interna foi mais freqüente no quartil superior de PCR-us, em 3173 pacientes com placas assintomáticas. Este resultado manteve se significativo somente nas mulheres, após ajuste para outros fatores de risco. No estudo de Rotterdam, os níveis de PCR-us, interleucina-6 e de moléculas de adesão foram comparados à gravidade da aterosclerose, em sítios arteriais distintos. Na bifurcação carotídea, somente níveis do tercil superior da PCR-us (? 0,28 g/dl) tiveram associação com espessamento médio-intimal, e duplicaram o risco de desenvolver-se estenose moderada ou grave (? 50%) [55]. A associação entre PCR-us, espessamento médio-intimal da carótida e risco de AVE foi prospectivamente avaliada em 5417 pacientes, no Cardiovascular Health Study [56]. Observou-se correlação fortemente positiva entre os níveis de PCR-us e AVE, nos casos com espessamento presente (p<0,02).
A associação entre concentração plasmática de PCR-us, estrutura histológica e imunocitológica da placa de carótida, e presença de sintomas neurológicos foi criteriosamente investigada por Alvarez-Garcia et al. [57], em 62 pacientes submetidos à EC. A média de PCR-us foi significativamente superior nos casos sintomáticos e nos classificados como instáveis à histologia (p<0,001), demonstrando-se também uma correlação positiva com a presença de magrófagos e linfócitos T na placa. Estes resultados corroboram o emprego da PCR-us como marcador de vulnerabilidade da doença carotídea. Entretanto, o fato do grupo classificado como clinicamente instável incluir também pacientes com quadros isquêmicos antigos, sem sintomas recentes, pode ser viés de confusão relevante e dificultar a interpretação dos resultados.
Por outro lado, a associação de PCR-us, interleucina-6 e infarto cerebral silencioso foi prospectivamente avaliada em 194 pacientes sem evidências de doença carotídea, coronariana ou arteriopatia periférica, no período de 10 anos. Nos 40 casos de infarto cerebral silencioso de pequena extensão, documentados por ressônancia encefálica, os níveis de PCR-us e IL-6 mostraram-se significativamente superiores, o que sugere uma ação pró-inflamatória destes mediadores em artérias intracranianas [58].
Entretanto, o comportamento da PCR-us em pacientes com doença coronariana e estenose carotídea tem sido descrito como não linear e controverso por alguns autores. Choi et al. [59] avaliaram, prospectivamente, o desenvolvimento de eventos coronarianos e espessamento médio-intimal da carótida interna, em uma coorte de 122 pacientes hipertensos, e compararam com os níveis de interleucina-6 e PCR-us. Além de a PCR-us apresentar-se significativamente elevada somente nos casos de doença coronariana, este marcador não obteve correlação com doença de carótidas, nem mesmo quando comparado a controles normotensos.
Recentemente, a American Heart Association publicou revisão sistemática com as recomendações para uso clínico de vários marcadores inflamatórios na aterosclerose. Em relação à PCR-us, concluiu-se que não encontrava-se justificado ainda seu emprego como preditor isolado de eventos na população geral, e sim como adjuvante em pessoas com risco estimado de doença coronária entre 10% a 20%, em 10 anos [60]. Entretanto, a extensão deste guidelines correspondente às doenças cerebrovasculares demonstrou haver evidência insuficiente para o uso sistemático da PCR-us na prevenção primária do AVE. Na prevenção secundária, há reconhecido valor prognóstico quando associada a outros fatores, e seu papel como determinante de decisões terapêuticas ainda não está estabelecido [61].
A influência da dosagem de PCR-us associada aos níveis séricos do complexo fosfolipase A2-lipoproteína, na incidência de AVE, foi recentemente investigada no estudo ARIC, em 12.819 pacientes. Após ajuste para outros fatores de risco, quando PCR-us e complexo fosfolipase A2-lipoproteína encontravam-se nos tercis superiores, o risco de AVE elevou-se oito vezes (p<0,001) [62]. Resultados similares foram relatados no Rotterdam study, que analisou a relação entre complexo fosfolipase A2-lipoproteína e desenvolvimento de doença coronária e/ou AVE [63].
O fator quimiotático de monócitos 1 tem sido referido como relacionado à dislipidemia e à instabilização da doença coronária, por alguns autores [64,65]. No Dallas Heart Study [66], a proporção de pacientes com coronariopatia detectável aumentou linearmente conforme a elevação dos quartis de fator quimiotático de monócitos 1 (de 17% no quartil inferior para 32% no quartil maior - p<0,0001). Expressão aumentada de fator quimiotático de monócitos 1 foi descrita em carótidas ateromatosas de humanos, em comparação às sadias, porém sua relação com desfechos agudos ainda não é clara [67].
Agentes infecciosos
A possível contribuição de alguns processos infecciosos, acelerando o crescimento das placas de ateroma, ou facilitando a ruptura e/ou instabilização, vem sendo bem estudada, em especial a infecção por Chlamydia pneumoniae (clamídia). Anticorpos contra clamídia têm sido detectados em espécimes de placas coronarianas e de carótida, o que sustenta a hipótese de causalidade [68,69]. Fagerberg et al. [70] estudaram, prospectivamente, a associação entre anticorpos contra clamidia e citomegalovírus com a incidência de eventos cerebrovasculares. Analisando 152 homens durante 6,5 anos, os autores demonstraram um risco oito vezes maior de AVE, nos pacientes com altos títulos contra clamídia, mas não contra citomegalovírus (p=0,043). Posteriormente, os mesmos pesquisadores observaram que a seropositividade para clamídia associava-se ao espessamento médio-intimal da carótida em 113 homens com hipertensão arterial [71].
A correlação entre infecção recente por clamídia e instabilidade clínica da doença carotídea foi investigada por Katsenis et al. [19], estudando 35 pacientes submetidos à EC. A positividade dos anticorpos se relacionou com a presença de sintomas, mas não com características ultra-sonográficas de vulnerabilidade. Gibbs et al. [72], em estudo mais criterioso, compararam a presença de clamídia na placa, o grau de embolização perioperatória (por Doppler transcraniamo) e a ocorrência de infarto cerebral em 98 pacientes sintomáticos. A constatação de que somente 25% dos casos tinham evidências de clamídia na placa e a ausência de correlação deste achado com taxa de embolização ou AVE ipsilateral sugerem que outros mecanismos estejam envolvidos no processo de instabilização. Controvérsia semelhante há em relação ao uso de antibióticos contra clamídia como estratégia de prevenção secundária e terciária na doença cerebrovascular. Ensaio clínico conduzido por Sander et al. [73] comparou o efeito da roxitromicina no desenvolvimento de placas de carótida em 272 pacientes acima de 55 anos, com infarto cerebral prévio. Após seguimento de três anos, no grupo intervido, houve diminuição significativa do espessamento médio-intimal, nos casos com titulação positiva, porém nenhuma alteração nos pacientes não reagentes. No entanto, Ieven et al. [74] não observaram diferença na titulação de anticorpos IgG contra clamídia, aferidos também por imunofluorescência, entre 106 pacientes com doença coronária, e 112 controles sadios, provavelmente por reação cruzada com outras bactérias. Por outro lado, Vainas et al. [75] descreveram associação entre seropositividade IgA, e tromboembolismo intra-operatório em placas consideradas histologicamente estáveis, sugerindo que a infecção por clamídia possa exercer também um efeito trombogênico na aterosclerose.
Estudo prospectivo recente, analisando o comportamento dos anticorpos contra Clamídia, herpes vírus e citomegalovírus, em 109 pacientes com estenose carotídea acima de 50%, não apontou diferenças na titulação entre os grupos, nem na comparação entre pacientes assintomáticos e sintomáticos, ou mesmo na comparação com os níveis de PCR-us, enfraquecendo a hipótese de que processos infecciosos deteminem diretamente a instabilização de lesões existentes [76].
Papel dos auto-anticorpos
O papel da ativação do sistema imune, a partir de anticorpos anticélulas endoteliais, vem sendo recentemente pesquisado na doença aterosclerótica. Farsi et al. [7], procedendo a dosagem de anticorpos anti-?2 glicoproteína (IgG e IgM - ELISA) em 93 pacientes submetidos à angioplastia coronária percutânea, e comparando a 105 controles, observaram que a prevalência dos anticorpos foi de 13% no grupo com doença coronária e de apenas 1% no grupo controle (p<0,001). Além disso, dentro do grupo coronariopata, os títulos foram significativamente maiores nos pacientes com angina instável do que os com angina estável (p<0,01), e a taxa de reestenose angiograficamente documentada foi de 66% nos casos com anticorpos positivos, e de somente 14% nos pacientes negativos (p<0,0004). Estes achados em conjunto sugerem a presença de um estado inflamatório imunoativado na doença instável, ou submetida à injúria endotelial por cateter.
A correlação dos níveis de anticorpos anticardiolipina com o risco de infarto agudo do miocárdio foi estudada em uma coorte do Helsinki Heart Study, que, entre 19.000 pacientes, acompanhou 133 homens portadores de dislipidemia por cinco anos. A dosagem de anticorpos da classe IgG foi significativamente maior nos casos que desenvolveram infarto subseqüente ou morte súbita do que nos controles (0,417 vs 0,361; p<0,005)[77]. Sherer et al. [78] também evidenciaram elevação de anticorpos anti-cardiolipina e anti-?2 glicoproteína em pacientes com doença coronária com e sem hipertensão arterial. Resultados semelhantes em relação à arteriopatia periférica foram escritos por de Godoy et al. [79], em 40 pacientes com idade entre 45 e 84 anos, nos quais o risco de desenvolver claudicação quadriplicou quando níveis elevados de anticorpos anticardiolipina foram observados (p<0,0001). Estudando a relação entre anticorpos anti-?2 glicoproteína e infarto cerebral, Staub et al. [80] compararam 92 casos de AVE com 93 controles, demonstrando que níveis elevados de anticorpos da classe IgA conferiram risco significativamente aumentado para a ocorrência do desfecho (p=0,025). Entretanto, análise recente de uma coorte de 4974 pacientes do estudo de Framingham [81] demonstrou que a elevação dos títulos de anticorpos anticardiolipina conferiu risco significativo de desenvolver AVE e/ou de AIT apenas nas mulheres.
Na doença obstrutiva de carótidas, já foi demonstrada por George et al. [82] expressão aumentada de anticorpos anti-?2 glicoproteína nas regiões subendoteliais da placa, possivelmente indicando interação com células endoteliais, macrófagos e linfócitos. Entretanto, somente estudos futuros poderão confirmar se os anticorpos anticardiolipina constituem fator de risco independente, ou meramente um fenômeno incidental, nos processos ateroscleróticos [10].
Também o papel citoprotetor das proteínas de choque térmico (heat shock protein) tem sido investigado no curso das doenças cardiovasculares. As proteínas de choque térmico constituem uma família de proteínas com peso molecular entre 20 e 150 daltons, que demonstram seqüências de aminoácidos altamente homogêneas entre espécies diferentes, como bactérias e o homem. Elas têm sua expressão aumentada nas situações de estresse, como infecções, alta temperatura, exposição a radicais livres e injúria endotelial. Várias evidências experimentais e clínicas têm relacionado a expressão das proteínas de choque térmico, ou mesmo os títulos de anticorpos, à gravidade e à instabilidade da doença aterosclerótica. Estas análises utilizam proteínas de choque térmico recombinantes de agentes infecciosos, como micobactérias, Helicobacter pylori e Chlamydia pneumoniae, atribuindo uma possível relação de eventos cardiovasculares agudos a uma resposta auto-imune desencadeada ou não pela exposição a bactérias [9,83-86].
Birnie et al. [87] pesquisaram a possível associação de anticorpos antiproteína de choque térmico 65 com aterosclerose coronária, em 136 pacientes submetidos a coronariografia. Estes autores constataram que as titulações do anticorpo se relacionaram com extensão e gravidade da doença, e que pacientes com infecção por Helicobacter pylori tratados de forma efetiva exibiram queda nas titulações.
Zhu et al. [88], por sua vez, avaliaram a possibilidade de que anticorpos contra proteína de choque térmico 60 humana conferirem risco para aterosclerose. Dos 391 pacientes submetidos a coronariografia, 75% apresentaram fração IgG positiva. A presença do anticorpo esteve também associada à gravidade da doença, pois títulos altos se relacionaram à doença em maior número de vasos, mesmo após o ajuste para outros fatores de risco.
Prohaszka et al. [89] avaliaram ambos anticorpos para proteína de choque térmico humana 60 e 65, em uma coorte envolvendo três grupos de pacientes: 1) coronariopatas graves submetidos à cirurgia de revascularização; 2) pacientes com fatores de risco para cardiopatia isquêmica e coronariografia normal; 3) controles sadios. Na análise multivariada, apenas os níveis de anticorpos para proteína de choque térmico 60 foram consideravelmente mais altos no grupo I do que nos outros grupos.
Mukherjee et al. [90] investigaram o papel dos anticorpos antiproteína de choque térmico em pacientes que evoluíram com reestenose de artérias coronárias, após angioplastia percutânea. O grupo de pacientes com reestenose cursou com níveis sustentados do anticorpo, enquanto aqueles com evolução favorável apresentaram queda nos seus títulos, sugerindo que um declínio nos níveis pode indicar um prognóstico melhor após revascularização.
Na doença obstrutiva de carótidas, a correlação com títulos elevados de anticorpos antiproteína de choque térmico 65 foi primeiramente demonstrada por Xu et al. [91]. Em estudo transversal populacional, os autores dosaram os títulos séricos de anticorpos IgG contra proteína de choque térmico 65 recombinante de micobactéria em 867 indivíduos com idade entre 40 e 79 anos e compararam os resultados com a avaliação da bifurcação carotídea por ultra-sonografia. Houve uma correlação significante entre títulos elevados de anticorpos e presença de placas nos pacientes acima de 60 anos, mesmo após controle de fatores como sexo, tabagismo, diabete e dislipidemia (p < 0,003).
O significado clínico desta associação foi investigado subseqüentemente pelos mesmos autores, por meio do seguimento de 750 pacientes arrolados para o estudo anterior, constatando-se que a elevação dos referidos anticorpos não somente apresentou forte correlação com lesões evolutivas de carótida (r = 0,78), como também foi um preditor independente de mortalidade em 5 anos (p<0,001) [92].
Somente por meio de estudos futuros, a relevância dos anticorpos antiproteína de choque térmico na aterogênese, e suas possíveis implicações diagnósticas e terapêuticas, poderão estar melhor compreendidas [93].
Aspectos morfológicos do ateroma na doença carotídea
Embora o porcentual de estenose carotídea defina, na atualidade, as decisões terapêuticas, os eventos cerebrovasculares são determinados por placas vulneráveis, que em parte dependem de características morfológicas distintas. Nesse contexto, vários estudos têm proposto aprimoramentos nas técnicas de imagem, no sentido de identificar as placas de alto risco, a maioria dos quais utilizando a composição histológica como padrão-ouro [11,94]. Entretanto, a correlação direta entre histologia e instabilidade da placa ainda não se encontra consistentemente estabelecida. Estudo clássico de Hatsukami et al. [95] analisou constituintes histológicos de 43 espécimes de EC, quanto à quantidade de fibrose intimal, lagos lipídicos ou necróticos, hemorragia intraplaca e calcificação, e comparou com a presença de sintomas pré-operatórios, não tendo encontrado diferenças significativas entre as classes clínicas. Recentemente, revisão sistemática de Lovett et al. [96] avaliou a qualidade e a comparabilidade de 73 estudos correlacionando exames de imagem e histologia da placa de carótida. Somente em 23% deles houve avaliação da reprodutibilidade na metodologia das diferentes técnicas de aquisição de imagem, e em apenas 12% os dados histológicos foram comparáveis, não havendo em nenhum dos estudos referência à possível limitação de resultados. Além de recomendarem importantes cuidados no acesso e leitura dos dados histológicos, como utilização de amostras representativas com número semelhante de casos com e sem sintomas, registro do tempo em relação aos sintomas, e do tempo entre a obtenção da imagem e da peça, cuidados de orientação espacial no corte e inclusão das peças, obtenção de cortes de, no máximo, 3 mm, e leitura cega, os autores reforçaram a necessidade de critérios mais homogêneos para estudos que visem avaliar a associação imagem-histologia.
Além de possuir papel definido na determinação do grau de estenose e de ser considerada suficiente para a decisão de intervenção, a ultra-sonografia tem contribuído substancialmente para a caracterização do conteúdo e da superfície da placa de carótida [97-101]. Estágios iniciais da aterosclerose podem ser monitorados pela espessura ultra-sonográfica da parede arterial, chamada espessamento médio-intimal, que, embora não defina se a infiltração é atribuída à íntima, ou por hipertrofia da média, é reconhecido marcador de evolução de lesões precoces [102]. Por outro lado, várias evidências indicam que o afilamento da capa fibrosa, a presença de conteúdo "mole", ou de ulceração, conferem instabilidade à placa [103,104], e que a proximidade dos "lagos" de necrose do lúmen, principalmente se associado à infiltração de macrófagos, predispõe a eventos clínicos [105]. Além disso, tanto em modelos utilizando a ultra-sonografia de superfície quanto em estudos mais recentes com reconstrução computadorizada ou ultra-sonografia intravascular, as imagens de baixa ecogenicidade têm sido relacionadas ao conteúdo necrótico ou hemorrágico da placa [106-108], e o grau de calcificação considerado inversamente proporcional à estabilidade das lesões [75]. É bem conhecido que, para estenoses de qualquer significado hemodinâmico, pacientes com placas menos ecogênicas apresentam maior incidência de infarto cerebral. Entretanto, a alta variabilidade intra e interobservador, na aquisição convencional de imagens e nos critérios de caracterização morfológica da placa, impôs aperfeiçoamentos técnicos [109]. Utilizando a reconstrução computadorizada da imagem, com estratificação em "escala de cinza", Gronholdt et al. [110] analisaram, prospectivamente, 146 pacientes com doença de carótida, durante quatro anos, e observaram que a baixa ecogenicidade associou-se a valores mais baixos na escala, e foi preditor independente de AVE, embora apenas no grupo de pacientes sintomáticos (n=135). Recentemente, Sztajzel et al. [111] acrescentaram o mapeamento de fluxo colorido à escala de cinza estratificada, em 28 pacientes submetidos à EC, e obtiveram forte correspondência histológica quanto à determinação de áreas hemorrágicas, lagos de necrose e irregularidade da superfície. Também a codificação da imagem do modo B, por um modelo computadorizado tridimensional, em tempo real, tem sido proposta na identificação das placas de risco, diminuindo artefatos ultra-sonográficos [112].
A ressonância nuclear magnética (RNM) de alta resolução representa outro expressivo avanço na análise morfológica das placas de ateroma, por meio da evolução de modelos inicialmente ex vivo, que se mostraram inefetivos, para estudos de correspondência histológica e clínica in vivo. Von Ingersleben et al. [113] compararam a imagem da bifurcação carotídea, obtida com a seqüência ponderada em T1, a cortes histológicos correspondentes, em pacientes submetidos à EC. Mesmo analisando apenas oito espécimes, já obtiveram significante correlação positiva na identificação de áreas de hemorragia, depósitos de lipídios, fibrose e calcificação. Posteriormente, o mesmo grupo testou a correspondência histológica das características da capa fibrosa - normal versus afilada versus rota - em 22 pacientes. Utilizando o mesmo protocolo de RNM, e considerando espessura ? 0,25 mm da capa fibrosa como normal, o coeficiente de correlação observado foi de 0,88 (p = 0,01) [114]. Em outro estudo, a ruptura da capa fibrosa foi fortemente associada à história recente de AIT ou AVE ipsilateral, e ocorreu com mais freqüência em placas com capa fibrosa adelgaçada, ou com grande centro lipídico [115].
A composição da placa pode ser determinada por RNM utilizando uma combinação de seqüências de pulsos, que variam conforme o tipo de estrutura que se pretende identificar, havendo por isto diferentes classificações propostas. Modelos baseados na quantificação de tecidos têm-se mostrado acurados e reprodutíveis [11]. Saam et al. [116] estudaram com RNM 31 candidatos à EC, utilizando seqüências com ponderações em T1, T2, e time-of-flight (TOF). Na quantificação de centros lipídicos ou necróticos, perda de matriz extracelular, calcificação e fibrose, obtiveram correlação histológica entre 73% e 95% dos casos, propondo a inclusão da técnica na avaliação prospectiva de placas ainda sem indicação de intervenção.
De particular interesse tem sido a detecção, pela RNM, de áreas de hemorragia ou de trombos murais, frequentemente associados a placas complexas e eventos clínicos. Moody et al. [117] observaram que a presença de metahemoglobina produz uma amplificação do sinal na sequência ponderada em T1, o que confere à área na qual existe hemorragia subaguda ou trombo recente um aspecto de sinal hiperintenso. Dentre 63 placas que apresentaram brilho intenso na RNM, 44 apresentaram pelo menos um dos quatro critérios histológicos de hemorragia, conferindo ao método uma especificidade de 84% e um valor preditivo positivo de 93%, com mínima variabilidade interobservador. Em estudo seguinte, os mesmos autores correlacionaram o sinal hiperintenso na placa, à presença de sintomas ipsilaterais recentes. Dados da RNM de 120 pacientes com AIT ou AVE foram comparados aos de 14 controles normais, e também à carótida contralateral (assintomática) do mesmo paciente. O sinal hiperintenso esteve presente em 60% das carótidas sintomáticas, em 36% das carótidas assintomáticas, e em nenhum dos controles (p < 0,001) [118]. A hipótese de que a hemorragia intraplaca estimula a progressão de placas existentes foi testada por Takaya et al. [119], em 29 pacientes acompanhados por 18 meses. Neste período, a média de aumento do volume parietal, e do volume dos centros lipídicos, respectivamente, foi de 7% e 28% nos 14 casos com hemorragia intraplaca ao início do estudo, e de 0,15% e 5%, nos controles sem hemorragia prévia (p < 0,01). Além disso, novas áreas de hemorragia foram observadas em 43% dos casos e em nenhum dos controles (p = 0,006). A Figura 1 demonstra exemplos de angiografias por RNM, com e sem sinal hiperintenso.
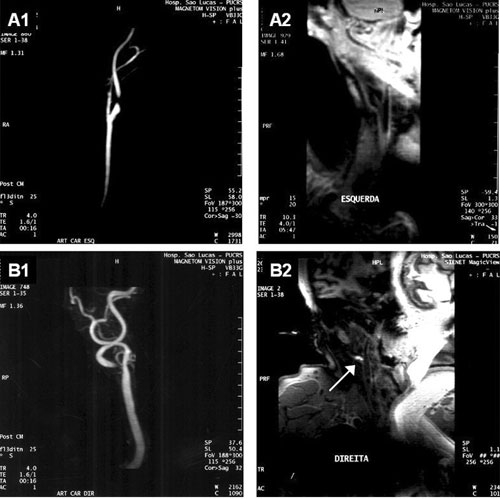
Fig. 1 - A1) Paciente assintomático com estenose de 90%; A2) ausência de sinal hiperintenso na seqüência ponderada em T1; B1) paciente com amaurose fugaz há uma semana, e estenose de 90%; B2) sinal hiperintenso demonstrando hemorragia intraplaca na carótida interna (seta)
A determinação do tempo de evolução de hemorragias presentes na placa carotídea, pela RNM de alta resolução, foi recentemente proposta por Chu et al. [120]. Em 27 pacientes submetidos a RNM com seqüências ponderadas em T1, T2, e densidade de prótons (PDW), a qualificação da hemorragia em fresca, recente, ou antiga, demonstrou forte correlação histológica (coeficiente kappa = 0,7) e boa acurácia (especificidade de 90% e sensibilidade de 74%). Além disso, os autores sugerem que a utilização desta seqüência na RNM seja capaz de definir a precisa localização dos trombos em lesões avançadas, diferenciando hemorragia na placa de trombos justa-luminais [121]. Uma vez que a hemorragia intraplaca, sem ruptura da capa fibrosa, não tem comprovada sua relação direta com sintomas, da mesma forma que trombos adjacentes à luz em geral indicam erosão, ulceração ou ruptura da placa, essa capacidade de diferenciação pela RNM, se comprovada em estudos subseqüentes, poderá ter importante implicação terapêutica no contexto da vulnerabilidade.
Ainda em avaliação preliminar, a RNM intravascular parece ser uma promissora técnica de caracterização da placa, principalmente da superfície intimal, com correspondência histológica descrita em modelos ex vivo [122]. Da mesma forma, um aprimoramento recentemente testado in vitro por Clarke et al. [123], diz respeito à validação de algoritmos automáticos de classificação da placa por RNM, utilizando como parâmetro a graduação histopatológica da American Heart Association [124].
Futuramente, técnicas como o imageamento molecular da atividade dos macrófagos, de mensuração da angiogênese, da atividade enzimática intraplaca, da apoptose celular, e de partículas-alvo da atividade inflamatória deverão incorporar o cenário dos métodos de imagem na aterosclerose [9].
Na atualidade, os exames de uso corrente, na caracterização da placa de carótida, encontram-se descritos na Tabela 1.
CONCLUSÃO
A aterosclerose de artérias carótidas tem aquirido grande relevância epidemiológica, pela alta prevalência na população idosa, e por ser causa freqüente de AVE. A endarterectomia cirúrgica se constitui o tratamento de escolha nas lesões graves, tendo sua indicação referendada hoje apenas pelo porcentual de estenose.
Entretanto, o emergente conceito de vulnerabilidade da placa, incorporado a partir do conhecimento de vários mecanismos bioquímicos, metabólicos, inflamatórios, e autoimunes, presentes nas diferentes fases da aterogênese, tem estimulado o estudo de marcadores capazes de identificar as lesões de risco. Dentre os mediadores inflamatórios de expressão sistêmica, a PCR-us tem-se destacado como preditor da presença e da instabilidade de placas de carótida, embora interesse recente exista em relação ao comportamento do CD40, das metaloproteinases de matriz extracelular, do complexo fosfolipase A2-lipoproteína. O papel dos anticorpos anticardiolipina e antiproteínas de choque térmico na doença de carótida, a despeito de vários estudos experimentais e clínicos, permanece incerto, assim como a suposta relação de causalidade entre infecção por clamídia e aterosclerose.
No âmbito dos métodos de imagem, a caracterização morfológica da placa tem sido prioritária no aprimoramento tanto da ultra-sonografia quanto da RNM. A hemorragia intraplaca, áreas hipocelularizadas de conteúdo rico em lipídios, afilamento e/ou ruptura da capa fibrosa tem sido relacionados como sinalizadores de risco iminente de eventos, principalmente através da RNM de alta resolução.
Por todos estes fatores, uma nova estratificação de risco aterotrombótico na doença carotídea deverá envolver, no futuro próximo, a combinação de marcadores inflamatórios sistêmicos, de imagem de alta resolução por RNM, de medidas da atividade molecular intraplaca, com o já consagrado porcentual angiográfico de estenose, possivelmente redefinindo os critérios atuais de intervenção.
REFERÊNCIAS
1. Kullo IJ, Edwards WD, Schwartz RS. Vulnerable plaque: pathobiology and clinical implications. Ann Intern Med. 1998;129(12):1050-60.
2. Ross R. Atherosclerosis: an inflammatory disease. N Engl J Med. 1999;340(2):115-26.
3. Ballantyne CM, Nambi V. Markers of inflammation and their clinical significance. Atheroscler Suppl. 2005;6(2):21-9.
4. Leinonen M. Pathogenic mechanisms and epidemiology of Chlamydia pneumoniae. Eur Heart J. 1993;14(suppl. K):57-61.
5. Grayston JT. Chlamydia in atherosclerosis. Circulation. 1993;87(4):1408-9.
6. Katsenis C, Kouskouni E, Kolokotronis L, Rizos D, Dimakakos P. The significance of Chlamydia pneumoniae in symptomatic carotid stenosis. Angiology. 2001;52(9):615-9.
7. Farsi A, Domeneghetti MP, Brunelli T, Gori AM, Fedi S, Gensini GF et al. Activation of the immune system and coronary artery disease: the role of anti-endothelial cell antibodies. Atherosclerosis. 2001;154(2):429-36.
8. Wick G, Xu Q. Atherosclerosis: an autoimmune disease. Exp Gerontol. 1999;34(4):559-66.
9. Mandal K, Jahangiri M, Xu Q. Autoimmunity to heat shock proteins in atherosclerosis. Autoimmun Rev. 2004;3(2):31-7.
10. Sherer Y, Shoenfeld Y. Antiphospholipid antibodies: are they pro-atherogenic or an epiphenomenon of atherosclerosis? Immunobiology. 2003;207(1):13-6.
11. Nighoghossian N, Derex L, Douek P. The vulnerable carotid artery plaque. Current imaging methods and new perspectives. Stroke. 2005;36(12):2764-72.
12. Biasucci LM. CDC/AHA Workshop on markers of inflammation and cardiovascular disease. Application to clinical and public health practice. Clinical use of inflammatory markers in patients with cardiovascular diseases. A background papper. Circulation. 2004;110(25):e560-7.
13. Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336(14):973-9.
14. Waddington EI, Croft KD, Sienuarine K, Latham B, Puddey, IB. Fatty acid oxidation products in human atherosclerotic plaque: an analysis of clinical and histopatological correlates. Atherosclerosis. 2003;167(1):111-20.
15. Nishi K, Itabe H, Uno M, Kitazato KT, Horigushi H, Shinno K et al. Oxided LDL in carotid plaques and plasma associates wtih plaque instability. Arterioscler Thromb Vasc Biol. 2002;22(10):1649-54.
16. Johnsen SH, Mathiesen EB, Fosse E, Joakimsen O, Stensland-Bugge E, Njolstad I et al. Elevated high-density lipoprotein cholesterol levels are protective against plaque progression: a follow-up study of 1952 persons with carotid atherosclerosis the Tromso study. Circulation. 2005;112(14):498-504.
17. Olsson AG, Schwartz GG, Szarek M, Sasiela WJ, Ezekowitz MD, Ganz P et al. High-density lipoprotein, but not low-density lipoprotein cholesterol levels influence short-term prognosis after acute coronary syndrome: results from MIRACL trial. Eur Heart J. 2005;26(9):890-6.
18. Tirschwell DL, Smith NL, Heckbert SR, Lemaitre RN, Longstreth WT Jr., Psaty BM. Associaton of cholesterol with stroke risk varies in subtypes and patients subgroups. Neurology. 2004;63(10):1868-75.
19. Schönbeck U, Libby P. CD40 signaling and plaque instability. Circ Res. 2001;89(12):1092-103.
20. Schönbeck U, Varo N, Libby P, Buring J, Ridker PM. Soluble CD40L and cardiovascular risk in women. Circulation. 2001;104(19):2266-8.
21. Kinlay S, Schwartz GG, Olsson AG, Rifai N, Sasiela WJ, Szarek M et al. Effect of atorvastatin on risk of recurrent cardiovascular events after an acute coronary syndrome associated with high soluble CD40 ligand in the Myocardial Ischemia Reduction with Aggresssive Chosleterol Lowering MIRACL study. Circulation. 2004;110(4):386-91.
22. Blake GJ, Ostfeld RJ, Yucel EK, Varo N, Schönbeck U, Blake MA et al. Soluble CD40 ligand levels indicate lipid accumulation in carotid atheroma: an in vivo study with high-resolution MRI. Arterioscler Thromb Vasc Biol. 2003;23(1):e11-4.
23. Sapienza P, di Marzo L, Borrelli V, Sterpetti A, Mingoli A, Cresti S et al. Metalloproteinases and their inhibitors are markers of plaque instability. Surgery. 2005;137(3):355-63.
24. Loftus IM, Naylor AR, Bell PR, Thompson MM. Plasma MMP-9: a marker of carotid plaque instability. Eur J Vasc Endovasc Surg. 2001;21(1):17-21.
25. Molloy KJ, Thompson MM, Jones JL, Schwalbe EC, Bell PR, Naylor AR et al. Unstable carotid plaque exhibit raised matrix metalloproteinase-8 activity. Circulation. 2004;110(3):337-43.
26. Sluijter JP, Pulskens WP, Schoneveld AH, Velema E, Stridjer CF, Moll F et al. Matrix metalloproteinase 2 is associated with stable and matrix metalloproteinases 8 and 9 with vulnerable carotid atherosclerotic lesions: a study in human endarterectomy specimen pointing to a role for different extracellular matrix metalloproteinase inducer glycosylation forms. Stroke. 2006;37(1):235-9.
27. Verhoeven B, Hellings WE, Moll FL, de Vries JP, de Kleijn DP, de Bruin P et al. Carotid atherosclerotic plaque in patients with transient ischemic attacks and strokes have unstable characteristics compared with plaques in asymptomatic and amaurosis fugax patients. J Vasc Surg. 2005;42(6):1075-81.
28. Von der Thüsen JH, Kuiper J, Van Berkel T, Biessen EA. Interleukins in atherosclerosis: molecular pathways and therapeutic potential. Pharmacol Rev. 2003;55(1):133-66.
29. Mallat Z, Corbaz A, Scoazec A, Besnard S, Lesèche G, Chvatchko Y et al. Expression of Interleukin-18 in human atherosclerotic plaque and relation to plaque instability. Circulation. 2001;104(14):1598-603.
30. Fantuzzi G, Reed DA, Dinarello CA. IL-12 induced IFN-gamma is dependent on caspase-1 processing of the IL-18 precursor. J Clin Invest. 1999;104(6):761-7.
31. Yamagami H, Kitagawa K, Hoshi T, Furukado S, Hougaku H, Nagai Y et al. Association of serum IL-18 levels with carotid intima-media thickness. Arterioscler Thromb Vasc Biol. 2005;25(7):1458-62.
32. Elkind MS, Rundek T, Sciacca RR, Ramas R, Chen HJ, Boden-Abdala B et al. Interleukin-2 levels are associated with carotid artery intima-media thickness. Atherosclerosis. 2005;180(1):181-7.
33. Mallat Z, Heymes C, Ohan J, Faggin E, Lesèche G, Tedgui A. Expression of interleukin-10 in advanced human atherosclerotic plaques. Relation to inducible nitric oxide syntethase expression and cell death. Arterioscler Thromb Vasc Biol. 1999;19(3):611-6.
34. Jialal I, Devaraj S, Venugopal SK. C-reactive protein: risk marker or mediator in atherothrombosis? Hypertension. 2004;44(1):6-11.
35. Koenig W. Predicting risk and treatment benefit in atherosclerosis: the role of C-reactive protein. Int J Cardiol. 2005;98(2):199-206.
36. Fu T, Borensztajn J. Macrophage uptake of LDL bound to aggregated C-reactive protein: possible mechanism of foam-cell formation in atherosclerotic lesions. Biochem J. 2002;366(pt 1):195-201.
37. Pasceri V, Willerson JT, Yeh ET. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation. 2000;102(18):2165-8.
38. Nakagomi A, Freedman SB, Geczy CL. Interferon-gamma and lipopolysaccharide potentiate monocyte tissue factor induction by C-reactive protein: relationship with age, sex and hormone replacement treatment. Circulation. 2000;101(15):1785-91.
39. Mold C, Gewurz H, Du Clos TW. Regulation of complement activation by C-reactive protein. Immunopharmacology. 1999;42(1-3):23-30.
40. Nakajima T, Schulte S, Warrington KJ, Kopecky SL, Frye RL, Goronzy JJ et al. T-cell-mediated lysis of endotelial cells in acute coronary syndromes. Circulation. 2002;105(5):570-5.
41. Venugopal SK, Devaraj S, Yuhanna I, Shaul P, Jialal I. Demonstration that C-reactive protein decrease eNOS expression and bioactivity in human aortic endothelial cells. Circulation. 2002;106(12):1439-41.
42. Morrow DA, Rifai N, Antman EM, Weiner DL, McCabe CH, Cannon CP et al. C-reactie protein is a potent predictor of mortality independently of and in combination with troponin T in acute coronary syndromes: a TMI IIA substudy. Thrombolysis in myocardial infarction. J Am Coll Cardiol. 1998;31(7):1460-5.
43. Heeschen C, Hamm CW, Bruemmer J, Simoons ML. Predictive value of C-reactive protein and troponin T in patients with unstable angina: a comparative analysis. CAPTURE investigators. J Am Coll Cardiol. 2000;35(6):1535-42.
44. Ridker PM, Rifai N, Pfeffer MA, Sacks FM, Moye LA, Goldman S et al. Inflammation, pravastatin, and the risk of coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and recurrent events (CARE) investigators. Circulation. 1998;98(9):839-44.
45. Retterstol L, Eikvar L, Bohn M, Bakken A, Erikssen J, Berg K. C-reactive protein predicts death in patients with previous premature myocardial infarction - a 10 year follow-up study. Atherosclerosis. 2002;160(2):433-40.
46. Tomoda H, Aoki N. Prognostic value of C-reactive protein levels within six hour after the onset of acute myocardial infarction. Am Heart J. 2000;140(2):324-8.
47. Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347(20):1557-65.
48. Albert MA, Glynn RJ, Ridker PM. Plasma concentration of C-reactive protein and calculated Framingham Coronary Heart Disease Score. Circulation. 2003;108(2):161-5.
49. Albert CM, Ma J, Stampfer MJ, Ridker PM. Prospective study of C-reactive protein, homocysteine and plasma lipid levels as predictors of sudden cardiac death. Circulation. 2002;105(22):2574-6.
50. Rost NS, Wolf PA, Kase CS, Kelly-Haes M, Silbershatz H, Massaro JM et al. Plasma concentration of C-reactive protein and risk of ischemic stroke and transient ischemic attack: the Framingham study. Stroke. 2001;32(11):2575-9.
51. Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Plasma concentration of C-reactive protein and risk of developing peripheral vascular disease. Circulation. 1998;97(5):425-8.
52. Kocer A, Canbulat C, Gozke E, Ilhan A. C-reactive protein is an indicator for fatal outcomes in first-time stroke patients. Med Sci Monit. 2005;11(11):540-4.
53. Arenillas JF, Alvarez-Sabin J, Molina CA, Chacon P, Montaner J, Rovira A et al. C-reactive protein predicts further ischemic events in first-ever transient ischemic attack or stroke patients with intracranial large-artery occlusive disease. Stroke. 2003;34(10):2463-8.
54. Wang TJ, Nam BH, Wilson PW, Wolf PA, Levy D, Polak JF et al. Association of C-reactive protein with carotid atherosclerosis in men and women: the Framingham Heart Study. Arterioscler Thromb Vasc Biol. 2002;22(10):1662-7.
55. van der Meer IM, de Maat MP, Bots ML, Breteler MM, Meijer J, Kiliaan AJ et al. Inflammatory mediators and cell adhesion molecules as indicators of severity of atherosclerosis. The Rotterdam Study. Arterioscler Thromb Vasc Biol. 2002;22(5):838-42.
56. Cao JJ, Thach C, Manolio TA, Psaty BM, Kuller LH, Chaves PH et al. C-reactive protein, carotid intima-media thickness, and incidence of ischemic stroke in the elderly: the Cardiovascular Health Study. Circulation. 2003; 108(2):166-70.
57. Alvarez-Garcia B, Ruiz C, Chacon P, Sabin JÁ, Matas M. High-sensitivity C-reactive protein in high-grade carotid stenosis: risk marker for unstable carotid plaque. J Vasc Surg. 2003;38(5):1018-24.
58. Hoshi T, Kitagawa K, Yamagami H, Furukado S, Hougaku H, Hori M. Relations of serum high-sensitivity C-reactive protein and interleukin-6 levels with silent brain infarction. Stroke. 2005;36(4):768-72.
59. Choi H, Cho DH, Shin HH, Park JP. Association of high-sensitivity C-reactive protein with coronary heart disease prediction, but not with carotid atherosclerosis, in patients with hypertension. Circ J. 2004;68(4):297-303.
60. Pearson TA, Mensah GA, Alexander RW, Anderson, JL, Cannon RO 3rd, Criqui M et al. Markers of inflammation and cardiovascular disease: application to clinical and public health pratice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107(3):499-511.
61. Di Napoli M, Schwaninger M, Cappelli R, Ceccarelli E, Gianfilippo G, Donati C et al. Evaluation of C-reactive protein measurement for assessing the risk and prognosis in ischemic stroke. A statement for health care professionals from the CRP Pooling Project Members. Stroke. 2005;36(6):1316-29.
62. Ballantyne CM, Hoogeveen RC, Bang H, Coresh J, Folsom AR, Heiss G et al. Lipoprotein-associated phospholipase A2, high-sensitivity C-reactive protein, and risk for incident stroke in middle-aged men and women in tha Atherosclerosis Risk in Communities (ARIC) study. Arterioscler Thromb Vasc Biol. 2004;24:E-127-8.
63. Oei HH, van der Meer IM, Hofman A, Koudstaal PJ, Stijnen T, Breteler MM et al. Lipoprotein-associated phospholipase A2 activity is associated with risk of coronary heart disease and ischemic stroke. The Rotterdam study. Circulation. 2005;111(5):570-5.
64. Kowalski J, Okopien B, Madej A, Makowiecka K, Zielinski M, Kalina Z et al. Levels of sICAM-1, sVCAM-1 and MCP-1 in patients with hyperlipoproteinemia IIa and IIb. Int J Clin Pharmacol Ther. 2001;39(2):48-52.
65. Matsumori A, Furukawa Y, Hashimoto T, Yoshida A, Ono K, Shioi T et al. Plasma levels of the monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 are elevated in patients with acute myocardial infarction. J Mol Cell Cardiol. 1997;29(1):419-23.
66. Deo R, Khera A, McGuire DK, Murphy SA, Meo Neto JP, Morrow DA et al. Association among plasma levels of monocyte chemoattractant protein-1, traditional cardiovascular risk factors, and subclinical atherosclerosis. J Am Coll Cardiol. 2004;44(9):1812-8.
67. Nelken NA, Coughlin SR, Gordon D, Wilcox JN. Monocyte chemoattractant protein-1 in human atheromatous plaques. J Clin Invest. 1991;88(4):1121-7.
68. Linnanmäki E, Leinonen M, Mattila K, Neiminen MS, Valtonen V, Saikku P. Chlamydia pneumoniae-specific circulating immune complexes in patients with chronic coronary heart disease. Circulation. 1993;87(4):1130-4.
69. Cochrane M, Walker P, Gibbs H, Timms P. Multiple genotypes of Chlamydia pneumoniae identified in human carotid plaque. Microbiology. 2005;151(pt 7):2285-90.
70. Fagerberg B, Gnarpe J, Gnarpe H, Agewall S, Wikstrand J. Chlamydia pneumoniae but not cytomegalovirus antibodies are associated with future risk of stroke and cardiovascular disease: a prospective study in middle-aged to elderly patients with treated hypertension. Stroke. 1999;30(2):299-305.
71. Schmidt C, Hulthe J, Wikstrand J, Gnarpe H, Gnarpe J, Agewall S et al. Chlamydia pneumoniae seropositivity is associated with carotid artery intima-media thickness. Stroke. 2000;31(7):1526-31.
72. Gibbs RG, Sian M, Mitchell AW, Greenhalgh RM, Davies AH, Carey N. Chlamydia pneumoniae does not influence atherosclerotic plaque behavior in patients with established carotid artery stenosis. Stroke. 2000;31(12):2930-5.
73. Sander D, Winbeck K, Klingelhofer J, Etgen T, Conrad B. Reduced progression of early carotid atherosclerosis after antibiotic treatment and Chlamydia pneumoniae seropositivity. Circulation. 2002;106(19):2428-33.
74. Ieven MM, Hoymans VY, Bosmans JM. Early carotid atherosclerosis and Chlamydia pneumoniae seropositivity: are there arguments to treat with antibiotics? Circulation. 2004;110(7):e74-5.
75. Vainas T, Kurvers HA, Mess WH, de Graaf R, Ezzahiri R, Tordoir JH et al. Chlamydia pneumoniae serology is associated with thrombosis-related but not with plaque-related microembolization during carotid endarterectomy. Stroke. 2002;33(5):1249-54.
76. Muller BT, Huber R, Henrich B, Adams O, Berns G, Siebler M et al. Chlamydia pneumoniae, herpes simplex vírus and cytomegalovirus in symptomatic and asymptomatic high-grade internal carotid artery stenosis. Does infection influence plaque stability? Vasa. 2005;34(3):163-9.
77. Vaarala O, Mänttäri M, Manninen V, Tenkanen L, Puurunen M, Aho K et al. Anti-cardiolipin antibodies and risk of myocardial infarction in a prospective cohort of middle-aged men. Circulation. 1995; 91(1):23-7.
78. Sherer Y, Tenenbaum A, Praprotnik S, Shemesh J, Blank M, Fisman EZ et al. Autoantibodies to cardiolipin and beta2-glycoproteinI in coronary artery disease patients with and without hypertension. Cardiology. 2002;97(1):2-5.
79. Godoy JM, Batigália F, de Godoy MR, Brandão AC, Souza DR. Anticardiolipin antibodies as a risk factor of atherosclerosis in intermittent claudication. Angiology. 2004;55(4):357-9.
80. Staub HL, Norman GL, Crowther T, da Cunha VR, Polanczyk A, Bohn JM et al. Antibodies to the atherosclerotic plaque components beta2-glycoproteinI and heat-shock proteins as risk factors for acute cerebral ischemia. Arq Neuropsiquiatr. 2003;61(3B):757-63.
81. Janardhan V, Wolf PA, Kase CS, Massaro JM, D'Agostino RB, Franzblau C et al. Anticardiolipin antibodies and risk of ischemic stroke and transient ischemic attack: the Framingham cohort and off spring study. Stroke. 2004;35(3):736-41.
82. George J, Harats D, Gilburd B, Afek A, Levy Y, Schneiderman J et al. Immunolocalization of beta 2-glycoprotein1 to human atherosclerotic plaque: potential implications for lesions progression. Circulation. 1999;99(17):2227-30.
83. Birnei DH, Holmet ER, McKay IC, Hood S, McColl KE, Hillis WS. Association between antibodies to heat shock protein 65 and coronary atherosclerosis: possible mechanism of action of Helicobacter pylori and other bacterial infections in increasing cardiovascular risk. Eur Heart J. 1998;19(3):387-94.
84. Fong IW, Chiu B, Viira E, Tucker W, Wood H, Peeling RW. Chlamydial Heat shock protein-60 antibody and correlation with Chlamydia pneumoniae in atherosclerotic plaques. J Infect Dis. 2002;186(10):1469-73.
85. Xu Q. Role of heat shock proteins in atherosclerosis. Arterioscler Thromb Vasc Biol. 2002;22(10):1547-59.
86. Benjamin IJ, McMillan DR. Stress (heat shock) proteins: molecular chaperones in cardiovascular biology and disease. Circ Res. 1998;83(2):117-32.
87. Birnie DH, Holme ER, McKay IC, Hood S, McColl KE, Hillis WS. Association between antibodies to heat-shock protein 65 and coronary atherosclerosis. Possible mechanism of action of Helicobacter pylori and other bacterial infections in increasing cardiovascular risk. Eur Heart J. 1998;19(3):387-94.
88. Zhu J, Quyyumi AA, Rott D, Csako G, Wu H, Halcox J et al. Antibodies to human heat-shock protein 60 are associated with the presence and severity of coronary artery disease: evidence for an autoimmune component of atherogenesis. Circulation. 2001;103(8):1071-5.
89. Prohaszka Z, Duba J, Horvath L, Csaszar A, Karadi I, Skebeni A et al. Comparative study on antibodies to human and bacterial 60 kDa heat shock proteins in a large cohort of patients with coronary heart disease and healthy subjects. Eur J Clin Invest. 2001;31(4):285-92.
90. Mukherjee M, De Benedictis C, Jewit D, Kakkar VV. Association of antibodies to heat-shock protein-65 with percutaneous transluminal coronary angioplasty and subsequent restenosis. Thromb Haemost. 1996;75(2):258-60.
91. Xu Q, Willeit J, Marosi M, Kleindienst R, Oberhollezner F, Kiechl S et al. Association of serum antibodies to heat-shock protein 65 with carotid atherosclerosis. Lancet. 1993;341(8840):255-9.
92. Xu Q, Kiechl S, Mayr M, Metzler B, Egger G, Oberhollezner F et al. Association of serum antibodies to heat-shock protein 65 with carotid atherosclerosis: clinical significance determined in a follow-up study. Circulation. 1999;100(11):1169-74.
93. Mehta TA, Greenman J, Ettelaie C, Venkatasubramaniam A, Chetter IC, McCollum PT. Heat shock proteins in vascular disease: a review. Eur J Vasc Endovasc Surg. 2005;29(4):395-402.
94. Yuan C, Mitsumori LM, Beach KW, Maravilla KR. Carotid atherosclerotic plaque: noninvasive MR characterization and identification of vulnerable lesions. Radiology. 2001;221(2):285-99.
95. Hatsukami TS, Ferguson MS, Beach KW, Gordon D, Detmer P, Burns D et al. Carotid plaque morphology and clinical events. Circulation. 1997;28(1):95-100.
96. Lovett JK, Redgrave JN, Rothwell PM. A critical appraisal of the performance, reporting, and interpretation of studies comparing carotid plaque imaging with histology. Stroke. 2005;36(5):1091-7.
97. Reilly LM, Lusby RJ, Hughes L, Ferrell LD, Stoney RJ, Ehrenfeld WK. Carotid plaque histology using real-time ultrasonography: clinical and therapeutic implications. Am J Surg. 1983;146(2):188-93.
98. Langsfeld M, Gray-Weale AC, Lusby RJ. The role of plaque morphology and diameter reduction in the development of new symptoms in asymptomatic carotid arteries. J Vasc Surg. 1989;9(4):548-57.
99. Goes E, Janssens W, Maillet B, Freson M, Steyaert L, Osteaux M. Tissue characterization of atheromatous plaques: correlation between ultrasound image and histological findings. J Clin Ultrasound. 1990;18(8):611-7.
100. Droste DW, Karl M, Bohle RM, Kaps M. Comparison of ultrasonic and histopathological features of carotid artery stenosis. Neurol Res. 1997;19(4):380-4.
101. Holdsworth RJ, McCollum PT, Bryce JS, Harrinson DK. Symptoms, stenosis and carotid plaque morphology: is plaque morphology relevant? Eur J Vasc Endovasc Surg. 1995;9(1):80-5.
102. Pignoli P, Tremoli E, Poli A, Oreste P, Paoletti R. Intimal plus medial thickness of the arterial wall: a direct measurement with ultrasound imaging. Circulation. 1986;74(6):1399-406.
103. Golledge J, Greenhalgh RM, Davies AH. The symptomatic carotid plaque. Stroke. 2000;31(3):774-81.
104. Tegos TJ, Sohail M, Sabetai M, Robless P, Akbar N, Pare G et al. Echomorphologic and histopathologic characteristics of unstable carotid plaques. Am J Neuroradiol. 2000;21(10):1937-44.
105. Bassiouny HS, Davis H, Massawa N, Gewertz BL, Glagov S, Zarins CK. Critical carotid stenoses: morphologic and chemical similarity between symptomatic and asymptomatic plaques. J Vasc Surg. 1989;9(2):202-12.
106. Wolverson MK, Bashiti HM, Peterson GJ. Ultrasonic tissue characterization of atheromatous plaques using a high resolution real time scanner. Ultrasound Med Biol. 1983;9(6):599-609.
107. Gronholdt ML. Ultrasound and lipoproteins as predictors of lipid-rich, rupture-prone plaques in the carotid artery. Arterioscler Thromb Vasc Biol. 1999;19(1):2-13.
108. Nissen SE, Yock P. Intravascular ultrasound: novel pathophysiological insights and current clinical applications. Circulation. 2001;103(4):604-16.
109. Arnold JA, Modaresi KB, Thomas N, Taylor PR, Padayachee TS. Carotid plaque characterization by dupplex scanning. Observer error may undermine current clinical trials. Stroke. 1999;30(1):61-5.
110. Gronholdt ML, Nordestgaard BG, Schroeder TV, Vorstrup S, Sillesen H. Ultrasonic echolucent carotid plaques predict future strokes. Circulation. 2001;104(1):68-73.
111. Sztajzel R, Momjian S, Momjian-Mayor I, Murith N, Djebaili K, Boissard G et al. Stratified gray-scale median analysis and color mapping of the carotid plaque: correlation with endarterectomy specimen histology of 28 patiens. Stroke. 2005;36(4):741-5.
112. Kern R, Szabo K, Hennerici M, Meairs S. Characterization of carotid artery plaques using real-time compound B-mode ultrasound. Stroke. 2004;35(4):870-5.
113. von Ingersleben G, Schmiedl UP, Hatsukami TS, Nelson JA, Subramaniam DS, Ferguson MS et al. Characterization of atherosclerotic plaques at the carotid bifurcation: correlation of high-resolution MR imaging with histologic analysis: preliminary study. Radiographics. 1997;17(6):1417-23.
114. Hatsukami TS, Ross R, Polissar NL, Yuan C. Visualization of fibrous cap thickness and rupture in human atherosclerotic carotid plaque in vivo with high-resolution magnetic resonance imaging. Circulation. 2000;102(9):959-64.
115. Yuan C, Zhang SX, Polissar NL, Echelard D, Ortiz G, Davis JW et al. Identification of fibrous cap rupture with magnetic resonance imaging is highly associated with recent transient ischemic attack or stroke. Circulation. 2002;105(2):181-5.
116. Saam T, Ferguson MS, Yarnykh VL, Takaya N, Xu D, Polissar NL et al. Quantitative evaluation of carotid plaque composition by in vivo MRI. Arterioscler Thromb Vasc Biol. 2005;25(1):234-9.
117. Moody AR, Murphy RE, Morgan PS, Martel AL, Delay GS, Allder S et al. Characterization of complicated carotid plaque with magnetic resonance direct thrombus imaging in patients with cerebral ischemia. Circulation. 2003;107(24):3047-52.
118. Murphy RE, Moody AR, Morgan PS, Martel AL, Delay GS, Allder S et al. Prevalence of complicated carotid atheroma as detected by magnetic resonance direct thrombus imaging in patients with suspected carotid artery stenosis and previous acute cerebral ischemia. Circulation. 2003;107(24):3053-8.
119. Takaya N, Yuan C, Chu B, Saam T, Polissar NL, Jarvik GP et al. Presence of intraplaque hemorrhage stimulates progression of carotid atherosclerotic plaques: a high-resolution magnetic resonance imaging study. Circulation. 2005;111(21):2768-75.
120. Chu B, Kampschulte A, Ferguson MS, Kerwin WS, Yarnykh VL, O'Briean KD et al. Hemorrhage in the atherosclerotic carotid plaque: a high-resolution MRI study. Stroke. 2004;35(5):1079-84.
121. Kampschulte A, Ferguson MS, Kerwin WS, Polissar NL, Chu B, Saam T et al. Differentiation of intraplaque versus juxtaluminal hemorrhage/thrombus in advanced human carotid atherosclerotic lesions by in vivo magnetic resonance imaging. Circulation. 2004;110(20):3239-44.
122. Larose E, Yeghiazarians Y, Libby P, Yucel EK, Aikawa M, Kacher DF et al. Characterization of human atherosclerotic plaques by intravascular magnetic resonance imaging. Circulation. 2005;112(15):2324-31.
123. Clarke SE, Beletsky V, Hammond RR, Hegele RA, Rutt BK. Validation of automatically classified magnetic resonance images for carotid plaque compositional analysis. Stroke. 2006;37(1):93-7.
124. Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W Jr et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Atherosclerosis, American Heart Associaton. Circulation. 1995;92(5):1355-74.

 All scientific articles published at rbccv.org.br are licensed under a Creative Commons license
All scientific articles published at rbccv.org.br are licensed under a Creative Commons license













 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket