Avaliar o impacto das cardiopatias congênitas em crianças no crescimento de crianças com síndrome de Down (SD) e a sua recuperação pôndero-estatural após correção cirúrgica.
Estudo retrospectivo de portadores da SD, entre 1984 e 2007. Excluídos os mosaicos e/ou portadores de morbidades associadas (n=165). Calcularam-se os escores Z para peso (Zpn) e comprimento (Zen) no momento do nascimento. Nos pacientes submetidos à correção cirúrgica (n=60), os escores Zp e Ze foram calculados antes da intervenção cirúrgica e em períodos posteriores, em até cinco anos de pós-operatório (PO). Em relação à população geral, Zp e Ze
2,5 foram significativos para déficit de crescimento. Utilizado teste do Qui-Quadrado para verificar relação entre peso/estatura e idade no momento da cirurgia e teste T de Student para avaliar o momento em que ocorreu a recuperação PO (
< 0,05).
As médias de Zpn (n=162) e Zen (n=156) foram -0,95 ± 1,27 e -1,348 ± 1,02. Da amostra total (n=165), 65,5% (n=108) dos pacientes apresentavam doença cardíaca. Dentre os pacientes submetidos (n=60) à cirurgia cardíaca, Zp era inferior a -2,5 em 55% (n=33) e Ze, em 60% (n=36). Com seis meses de PO, 67,4% alcançaram Zp
2,5. Em um ano, 85,7% atingiram Ze
2,5. Dividindo este grupo por idade, na época da cirurgia, em tercis não ocorreu diferença.
Observou-se déficit pôndero-estatural em relação à população geral desde o nascimento, sendo maior nas crianças com cardiopatia de indicação cirúrgica. A recuperação PO ocorreu em seis meses para o peso e em um ano para a estatura, sem diferença quanto à idade no momento cirúrgico.
To evaluate the impact of congenital heart diseases in growth of children with Down syndrome (DS) and the weight-height recovery after surgical correction.
Retrospective study of the DS patients between 1984 and 2007. Excluding the mosaics and/or patients with associated morbidities (n=165). Calculated Z scores for weight (Zwb) and length (Zlb) at birth. Those patients submitted to surgical correction (n= 60) these scores (Zw/Zh) were evaluated before surgery and in subsequent periods to five years. Malnutrition was defined as weight/height Z-score
2.5. Used Chi-square test to verify the relation between weight/length and age at the time of surgery and Student T test to evaluate the postoperative (PO) time of recovery (
< 0.05).
Means Zwb (n = 162) and Zlb (n = 156) were -0.95 ± 1.27 and -1.348 ± 1.02. From the total data (n = 165), 65.5% (n = 108) presented heart disease. Those submitted to cardiac surgery (n = 60), Zw was below -2.5 in 55% (n = 33) and Zh in 60% (n = 36). After six months PO, 67.4% achieved Zw
2.5. In one year, 85.7% achieved Zh
2.5. Dividing this group by age in tertiles at time of surgery no difference was found.
We concluded that malnutrition common in children with DS since birth. DS children with congenital heart and surgical indication were smaller and lighter than those without or with mild disease. PO recovery occurred in 6 months for weight and one year for height, with no difference in the age at the time of surgery.
INTRODUÇÃO
A Síndrome de Down (SD) é uma anormalidade cromossômica caracterizada pela cópia extra de material genético do cromossomo 21, podendo ocorrer de forma total ou parcial. Foi assim denominada após descrição dos aspectos clínicos pelo médico inglês John Langdon Haydon Down, em 1866 [1]. A ocorrência mundial da SD é estimada em cerca de um a dois casos a cada 1000 nascimentos. No Brasil, a estimativa desta população estava em torno de 300 mil pelo censo de 2000 do Instituto Brasileiro de Geografia e Estatística (IBGE) [2].
A idade materna após 35 anos é um dos principais fatores contribuintes para a ocorrência da trissomia do 21. Alguns estudos mostram que a idade paterna avançada também é um fator contribuinte [3].
O fenótipo da SD é caracterizado por mais de 80 características. As manifestações fenotípicas mais comuns são a deficiência intelectual, a hipotonia muscular, a baixa estatura, as características faciais e malformações congênitas, especialmente as cardíacas [4]. A SD também se caracteriza por disfunções e doenças de vários órgãos. Essas características clínicas podem variar consideravelmente em número e gravidade [5]. O fenótipo da SD foi inicialmente atribuído à perda do balanceamento cromossômico. Essa hipótese foi enfraquecida pelo fato de que outras trissomias autossômicas não apresentam variabilidade fenotípica. Correlações entre o genótipo e o fenótipo de pacientes com trissomias parciais do cromossomo 21 indicam que a região restrita no 21q22.2 está relacionada às principais características clínicas da SD. Isso suporta a hipótese de dosagem/efeito do gene [6]. Em 2007, Yahya-Graison et al. [7] sugerem que a expressão exacerbada desses genes estaria mais relacionada ao fenótipo da SD.
A baixa estatura final é uma das principais características do processo de crescimento dos portadores da SD, sendo que esse déficit já se inicia no período pré-natal [8]. Após nascimento, essa velocidade é muito reduzida entre seis e 36 meses em ambos os sexos. A puberdade, em geral, ocorre um pouco antes que na população geral e está associada a um déficit desse crescimento [9]. Existe também alta prevalência do sobrepeso e da obesidade, particularmente na adolescência e na idade adulta.
O crescimento pôndero-estatural é um bom indicador de saúde durante a infância e a adolescência. O crescimento e a estatura final diferentes das crianças com SD exigem o uso de curvas de peso e estatura/crescimento específicas [10]. Se colocarmos essas crianças nos gráficos da população geral, isso poderá mascarar a detecção de doenças adicionais como o hipotiroidismo, a doença celíaca e as cardiopatias. Da mesma forma, sobrepeso ou obesidade incipiente não serão reconhecidos [11,12].
Na SD, as malformações cardíacas ocorrem em 40% a 50% dessas crianças e a mais frequente é o defeito de septo atrioventricular (30% a 60%), seguida do defeito do septo ventricular (cerca de 30 %). Outras cardiopatias são: a comunicação interatrial
ostium secundum (cerca de 10%), a persistência do canal arterial e a tetralogia de Fallot. Em torno de vinte anos de idade, podem ocorrer o prolapso da valva mitral associado ou não ao da valva tricúspide e o refluxo aórtico [13].
Nas cardiopatias congênitas, o grau de comprometimento pôndero-estatural depende do tipo e da repercussão hemodinâmica das mesmas [14]. As crianças com doença cardíaca congênita moderada a grave apresentam maior comprometimento quando comparadas àquelas com cardiopatia leve ou às sem doença cardíaca. O aprimoramento das técnicas cirúrgicas e métodos diagnósticos nas últimas décadas têm permitido corrigir defeitos cardíacos mais precocemente e com melhores resultados.
A recuperação pôndero-estatural desses pacientes no pós-operatório, dentro dos potenciais genéticos, gera questionamentos e estudos. O aumento da sobrevida e melhora na qualidade de vida dessa população estão relacionados à correção cirúrgica das cardiopatias congênitas. A possibilidade de recuperação de crescimento destes pacientes após correção cirúrgica é justamente o objetivo principal deste estudo.
MÉTODOS
Casuística
Este trabalho partiu de um estudo de coorte retrospectivo de 181 pacientes acompanhados no ambulatório de Síndrome de Down do Serviço de Pediatria da Irmandade da Santa Casa de São Paulo no Brasil (ISCSP), pela revisão de prontuários no período de 20 de agosto de 1984 a 19 de setembro de 2007. A escolha dos prontuários foi aleatória ou conforme disponibilidade do serviço de arquivo médico da Santa Casa. O presente estudo foi aprovado pelo Comitê de Ética em Pesquisa da ISCSP. Todos apresentavam trissomia 21, com diagnóstico da síndrome comprovado por cariótipo, sendo excluídos os mosaicos. Do total de prontuários analisados (n=181), foram excluídos os pacientes que apresentaram níveis séricos de tetraiodotironina total (T4) e/ou livre (T4L) e/ou triiodotironina (T3) alterados para a idade e/ou quadro clínico de hipotiroidismo e leucemia e/ou fatores não cardiogênicos que poderiam comprometer o ganho pôndero-estatural, ficando a amostra reduzida a 165 indivíduos.
A idade dos pacientes no momento do primeiro atendimento variou entre 0 e 200 meses, com média de 49,8 meses (± 72,1) e mediana de 6,6 meses. Da amostra total (n=165), foram selecionados os portadores de cardiopatia e destes os que tinham indicação cirúrgica. Posteriormente, dividimos este grupo de pacientes em tercis, considerando a idade no momento da cirurgia cardíaca. O primeiro grupo (G1) incluiu as crianças com idade até 7,56 meses no momento da correção cirúrgica. O segundo grupo (G2) incluiu os que tinham entre 7,56 e 19,49 meses e o terceiro grupo (G3), os que tinham idade superior ao grupo anterior. Todos os pacientes foram submetidos à avaliação cardiológica para definir se havia cardiopatia ou não.
Nos pacientes portadores de cardiopatia, esse diagnóstico foi comprovado por exame clínico, eletrocardiograma, radiografia de tórax, ecocardiograma com Doppler colorido e, em alguns casos, por estudo hemodinâmico. Em todos os pacientes da amostra, foram realizadas dosagens semestrais de hormônios tiroideanos e hemograma. Anualmente, a todos foram solicitadas avaliações otorrinolaringológicas e oftalmológicas e nos não colecistectomizados foram realizados exames de ultrassonografia abdominal.
A rotina antropométrica do Serviço inclui enfermagem própria deste ambulatório que realiza as medidas, respeitando técnicas acuradas para a quantificação da estatura e do peso. Este ambulatório também dispõe de equipamentos antropométricos próprios. Os dados do nascimento foram levantados por registros do nascimento e os dados posteriores pelo levantamento dos prontuários dos pacientes atendidos neste ambulatório da ISCSP. Os valores de peso estavam expressos em gramas, os de estatura/comprimento em centímetros e os de idade das crianças foram convertidos em meses. Considerando o peso e a estatura como variáveis de distribuição homogênea em torno da média, calculou-se o escore Z. Como a amplitude de variação em relação à média é o desvio padrão, este cálculo permitiu estabelecer o quão distante a estatura e o peso de uma determinada criança se encontravam da média da população geral. Foi possível, ainda, combinar os valores de ambos os sexos para compor um só grupo de estudo. Alguns programas computadorizados fazem este cálculo automaticamente, mas este pode ser obtido pelas seguintes fórmulas:

Utilizamos neste estudo o
software Epiinfo CDC para cálculo de escore Z. Em todos os pacientes com registro do peso e comprimento no nascimento, foram realizados os cálculos de escore Z no momento do nascimento (escore Zpn/ Zen). Nos que foram submetidos à operação cardíaca, estes escores foram calculados antes e após a intervenção cirúrgica (6 meses, 1 ano, 2, 5 e 10 anos).
Dessa forma, determinamos o comprometimento do crescimento pôndero-estatural e o tempo necessário para a recuperação do crescimento após o procedimento cirúrgico. A análise estatística estabelecida foi realizada, tendo como instrumento de apoio os
softwares: BrOffice 3.0, EpiData e EpiData Analysis, for Windows/2000 XP.
Em todos os pacientes que foram submetidos à operação cardíaca (n=60), os escores Z foram calculados para peso e estatura antes da intervenção e nos vários momentos do pós-operatório descritos acima. Os valores de variáveis contínuas foram expressos em médias e desvio padrão. Consideramos comprometimento do crescimento pôndero-estatural, valores de escore do peso ou da estatura menores que -2,5 desvios padrão, em relação à população geral. A recuperação do escore Z do peso e da estatura foi considerada quando ocorreu diferença estatisticamente significante. Para avaliarmos o momento em que ocorreu esta recuperação, utilizamos o teste de diferença de médias (teste T de Student).
Para avaliar se existiu relação entre a recuperação do peso ou da estatura com a idade no momento da operação e nos períodos posteriores a ela, utilizamos o teste do Qui-quadrado de Pearson. O intervalo de confiança aceito como normal foi de 95%, sendo considerado como diferença estatística quando o nível de significância foi de 5% (
P < 0,05).
RESULTADOS
Das 165 crianças selecionadas no ambulatório de Síndrome de Down do Serviço de Pediatria da Santa Casa de São Paulo no Brasil, a média da idade das mães e dos pais (expressa em anos) no momento do nascimento da criança foi, respectivamente, 34 ±6,9 e 36 ±7,03. A idade variou entre 16 e 50 anos para as mães e entre 20 e 55 para os pais, com mediana de 34 e 36 anos. Os escores Z do peso dessas crianças no nascimento (n=162) variaram entre -5,63 e +1,72, com média de -0,95 ± 1,2. Os escores Z da estatura/comprimento no nascimento (n=156) variaram entre -6,77 e +0,53, com média de -1,34 e desvio padrão de 1,02. O comprometimento pôndero-estatural destas crianças com SD, em relação à população geral, ocorreu desde o nascimento.
Dos 165 pacientes com SD, 34,55% (n=57) não tinham cardiopatia (Grupo 2) e 65,45% (n=108) apresentavam doença cardíaca. Do total da amostra (n=165), 51% (n=84) eram do gênero feminino e as idades variavam, no momento do primeiro atendimento no ambulatório de SD da Santa Casa de São Paulo, entre 0 e 200 meses, com média de 30,38 e mediana de 6,6 meses. As cardiopatias encontradas (n=108) em ordem decrescente de frequência foram: defeito do septo atrioventricular (DSAV; 36%/n=39), comunicação interventricular (CIV; 30,5%/n=33), comunicação interatrial (CIA; 17,5%/n=19), associações de cardiopatias (6,5%/n=7), persistência do canal arterial (PCA; 4,6%/n=5), tetralogia de Fallot (T4F; 2,7%/n=3), e outras (Figura 1).

Fig. 1- Prevalência dos tipos de cardiopatias congênitas no Grupo 1 (n=108); eixo vertical: percentual das cardiopatias; eixo horizontal: tipos de cardiopatias; Assoc- associações de uma ou mais cardiopatias (n=7); CIA- comunicação interatrial (n=19); CIV- comunicação interventricular (n=33); DSAV- defeito do septo atrioventricular (n=39); outros- outros tipos de cardiopatias; PCA- persistência do canal arterial (n=5) e T4F- Tetralogia de Fallot (n=3)
Das 108 crianças com cardiopatia, 63 (58,3%) tiveram indicação cirúrgica e as demais (n=45) apresentaram cardiopatia com leve ou nenhuma repercussão ou que evoluíram para resolução espontânea. Destas 63 crianças, apenas 60 foram submetidas à operação cardíaca, porque uma foi a óbito antes da correção cirúrgica, uma abandonou o acompanhamento no Serviço e os responsáveis pela outra recusaram a operação. A idade, no momento da correção cirúrgica da cardiopatia variou entre 1,5 e 168,1 meses, com média de 25 meses (± 34,6) e mediana de 10,7 meses. Destas crianças, 58,3% eram do gênero feminino. A média do escore Z do peso antes da operação foi de -2,62 ± 1,35 e a média do escore Z da estatura foi de -3,01 (± 1,83). Observou-se que 55% destas crianças (n=33) tinham escore Zp inferior a -2,5 e, em relação à estatura, 60% (n=36) tinham valores de escore inferiores a -2,5. Observa-se, portanto, o comprometimento do peso e da estatura nestas crianças com SD e cardiopatia com indicação cirúrgica, sendo este comprometimento mais intenso para a estatura.
Com seis meses (6m) após a correção cirúrgica (n=50) da cardiopatia, a média dos valores dos escores Z do peso e da estatura foram, respectivamente, -1,95 (±1,17) e - 3,04 (+/-6,09). Um ano (1a) após a cirurgia (n=40), esses valores de média de escore Z obtidos para o peso e estatura foram de -1,32 ± 1,14 e de -2,00 ± 1,36. Com dois anos (2a) de pós-operatório (n=34), esses valores de média do escore Z peso foram de -0,99 ± 1,20 e de -1,58 ± 1,16. Cinco anos (5a) após a intervenção cirúrgica (n= 26), as médias dos escores do peso foram de -0,568 ± 1,29 e -1,65 ± 1,05 (Figura 2). Dez anos (10a) após o procedimento cirúrgico (n=13), a média do escore Z do peso foi -0,10 ± 0,86 e do Z estatura foi de -1,55 ± 0,76.
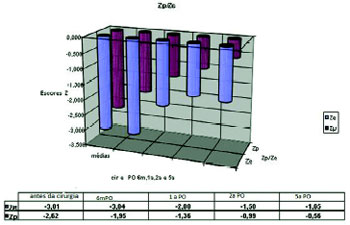
Fig. 2- Distribuição dos valores dos escores Z do peso e do comprimento (Zp/Ze) no momento da operação e períodos posteriores a ela; os valores dos escores Z estão expressos em médias
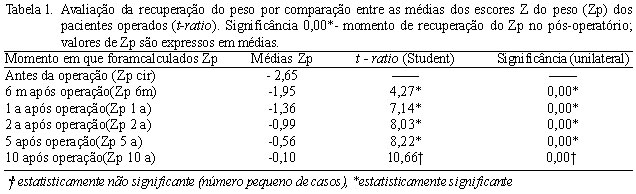
O teste T de Student foi utilizado para comparar as médias dos escores da estatura e do peso no momento da operação e nos períodos pós-operatórios. A recuperação do peso, após operação cardíaca, ocorreu na maioria desta população com SD seis meses após a operação, quando o escore Z do peso (Zp) alcançou média de -1,95 ± 1,17, 67,4% da amostra apresentaram um escore Zp acima de -2,5 e o T de Student foi 4,27 (Tabela 1).
Já a recuperação da estatura após a cirurgia ocorreu na maioria dos casos (85,7% com Zp acima de -2,5) em um ano de pós-operatório, quando o escore (Ze) alcançou uma média de - 2,0 ± 1,36, 85,7% destes pacientes alcançaram um Zp acima de -2,5 e o T de Student foi de 4,68 (Tabela 2).
A redução gradativa do número de casos ao longo do período de seguimento PO (n=60/ 6m PO:n=50/ 1a PO:n=40/ 2a PO: n=34/ 5a PO:n=26 e 10a PO:n=13) ocorreu por abandono do tratamento, pelo aparecimento de fatores não cardiogênicos (como hipotiroidismo e leucoses) que poderiam comprometer o crescimento e por óbitos. No primeiro semestre, a amostra inicial ficou reduzida para 50 casos porque a mortalidade foi maior (6,6 %/n=4) e seis casos foram excluídos (fatores acima relatados). Em dez anos de PO, o número de casos foi muito pequeno (n=13) e estatisticamente não significativo, sendo excluídos desta análise.
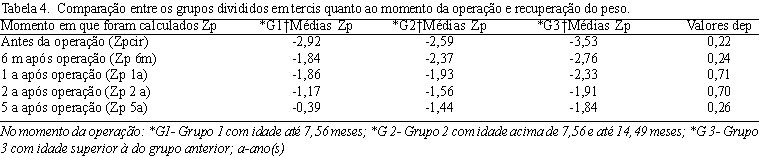
Posteriormente, dividiu-se o grupo por faixa etária na época da correção cirúrgica da cardiopatia em tercis. O primeiro grupo (Grupo 1) incluiu pacientes com idade de até 7,56 meses no momento da operação (33,3%/n=20); o segundo grupo (Grupo 2), pacientes com idade acima de 7,56 e até 19,49 (31,7%/n=19) e o terceiro grupo (Grupo 3) englobou os que tinham idade acima de 19,49 meses na época da correção cirúrgica (35%/n=21). O teste do Qui-Quadrado foi utilizado para verificar se a idade apresentava relação com o peso e a estatura no momento da operação e nos períodos a ela posteriores.
Após dez anos da operação, o número de pacientes não foi estatisticamente significativo, sendo estes casos excluídos da amostra. Os valores de
P foram calculados pelo teste do Qui-Quadrado (X
2) de Pearson para escores de peso e estatura no momento da cirurgia e nos períodos posteriores. Não houve diferença na recuperação tanto da estatura como do peso em relação à idade no momento da cirurgia (Tabelas 3 e 4). A mortalidade pós-operatória precoce foi maior nos pacientes do Grupo dois que apresentavam DSAV total e hipertensão arterial pulmonar secundária (15,7%/n=3). Todos foram previamente submetidos a cateterismo cardíaco antes da indicação cirúrgica e apresentavam testes de vasorreatividade pulmonar que permitiam esta correção.
DISCUSSÃO
O crescimento pôndero-estatural é um dos mais importantes indicadores de saúde da criança. As doenças crônicas acarretam comprometimento desse crescimento, sendo as cardiopatias congênitas um dos principais fatores determinantes desse comprometimento [15]. O desenvolvimento de novas técnicas cirúrgicas e o avanço da terapêutica farmacológica têm possibilitado maior sobrevida e melhora na qualidade de vida destes pacientes. O processo de recuperação do peso e da estatura após conduta terapêutica cirúrgica é foco habitual de preocupação dos pediatras, cardiopediatras e dos pais. Esse fato fez por merecer este estudo em particular numa população de crianças com SD. Nelas, a ocorrência de malformações cardíacas é frequente e existem características de crescimento e composição corporal inerentes a esta síndrome.
Entre os fatores de risco bem documentados para a ocorrência de não disjunções meióticas em trissomias, como a do cromossomo 21, está a idade materna avançada no momento do parto [16]. Mustacchi & Rozone [17] e outros autores já citados [3] também têm considerado a idade paterna avançada como variável contribuinte para a ocorrência da SD. Neste estudo, observamos correlação entre a ocorrência da SD tanto com a idade materna avançada no momento do parto como da idade paterna avançada.
São características do crescimento das crianças portadoras de SD: a baixa estatura final, a reduzida velocidade de crescimento e tendência à obesidade no final da infância e adolescência. O déficit pôndero-estatural das crianças com SD em relação à população geral se inicia no período pré-natal. Foi demonstrado neste estudo que o peso e comprimento eram menores desde o momento do nascimento, variando entre -0,9 e -1,3. Estes resultados foram compatíveis com os de Myrelid et al. [18] em que os desníveis encontrados no momento do nascimento variaram entre -1,0 e -1,5 para o comprimento e entre -1,2 e -1,5 para o peso. Observou-se certa divergência em relação aos resultados de Meguid et al. [19], em que estes desníveis foram maiores (-1,6 a -3,5DP) e em relação aos de Cronck [20], com menores desníveis (-0,5DP).
A ocorrência de malformações cardíacas nessa população é de cerca de 40 a 50%, sendo as mais frequentes o defeito do septo atrioventricular (DSAV) e a comunicação interventricular (CIV) [12,13]. Neste estudo, encontramos resultados semelhantes e as cardiopatias mais frequentemente encontradas foram o DSAV e a CIV. A incidência de meninas portadoras de cardiopatias (58,3%) foi um pouco maior em relação à dos meninos, como é descrito na literatura em amostras populacionais com SD [21].
Há consenso na literatura que, na população geral, a maioria das crianças com cardiopatias congênitas tem comprometimento de peso e/ou estatura, variando com a gravidade e com o tipo de cardiopatia. As cardiopatias com hiperfluxo pulmonar são as que cursam com comprometimento mais importante do peso que da estatura. Existem poucos estudos na literatura sobre o crescimento de crianças com SD portadoras de cardiopatias congênitas, ocorrendo controvérsias [18,22].
Neste estudo, observou-se comprometimento pôndero-estatural das crianças portadoras de cardiopatia com indicação cirúrgica, as quais, na sua maioria, apresentavam cardiopatias com hiperfluxo. Antes da operação, 55% (n=33) tinham o escore Zp inferior a -2,5 e, em relação ao escore Z da estatura (Ze), 60% (n=36) apresentavam valores inferiores a -2,5. O comprometimento da estatura foi maior que o do peso antes da operação.
A recuperação pôndero-estatural após correção cirúrgica da cardiopatia ocorre em todos os tipos de cardiopatias congênitas na população geral [23]. O grau de recuperação é controverso. São escassos os estudos realizados nesta área, envolvendo na sua maioria um seguimento curto de acompanhamento pós-operatório [24]. Na população geral, os estudos demonstram que o período pós-operatório necessário à recuperação pode variar entre seis meses e um ano [25], como na deste estudo. A recuperação do peso ocorreu na maioria dos casos após seis meses, quando 67,4% desta amostra apresentaram valores de escore Z do peso (Zp6m) acima de -2,5. Em relação à estatura, esta recuperação ocorreu na maioria dos casos (85,7% com valores acima de -2,5) com um ano de PO. Os valores de escores Z foram comparados aos da população geral porque não existem fórmulas específicas que utilizem as médias de peso/estatura da população com SD e nem curvas de crescimento específicas para crianças com SD e cardiopatia, sendo esta uma limitação do estudo.
Na população geral, a recuperação pôndero-estatural após operação cardíaca frequentemente é maior nos operados antes dos três anos de idade. Nesse grupo de pacientes, o comprometimento ponderal pré-operatório costuma ser maior, variando quanto ao tipo de cardiopatia [25]. Neste estudo, ao se dividir a amostra total em tercis quanto à idade no momento da operação, encontrou-se o mesmo padrão de recuperação. Nesta amostra de portadores de SD, observou-se discordância em relação à população geral quanto à idade no momento da correção cirúrgica da cardiopatia. Devemos levar em consideração que na população com SD a cardiopatia mais frequente (DSAV) é de hiperfluxo, acianótica e, na maioria dos casos, com indicação cirúrgica no primeiro ano de vida.
Em concordância com a literatura, as crianças do Grupo 2, que eram portadoras de DSAV total com evolução para doença vascular pulmonar, apresentaram mortalidade transoperatória aumentada. Recomenda-se que crianças com SD e DSAV total sejam submetidas à correção cirúrgica antes dos primeiros 12 meses de vida, preferencialmente entre três e seis meses de vida e antes do início da hipertensão arterial pulmonar.
CONCLUSÕES
As crianças com SD apresentam comprometimento do crescimento em relação à população geral desde o nascimento.
As cardiopatias congênitas com indicação cirúrgica determinaram maior comprometimento pôndero-estatural em 55% da amostra de pacientes com SD para o peso e em 60%, para a estatura.
A recuperação do peso e da estatura após correção cirúrgica desta população ocorreu na maioria dos casos em seis meses para o peso e, em um ano, para a estatura. Este comprometimento é mais acentuado para a estatura desde o nascimento.
Não ocorreu relação entre o ganho de peso no período pós-operatório e a idade no momento da operação.
AGRADECIMENTOS
Ao Conselho Nacional de Desenvolvimento e Pesquisa (CNPq) e à equipe do ambulatório de SD da Santa Casa de São Paulo.
REFERÊNCIAS
1. Dunn PM. Dr Langdon Down (1828-1896) and "mongolism". Arch Dis Child. 1991;66(7 Spec No):827-8.
2. IBGE (Instituto Brasileiro de Geografia e Estatística). Censo Demográfico 2000. Disponível em: (27 fev 2009).
3. Zhu JL, Madsen KM, Vestergaard M, Olesen AV, Basso O, Olsen J. Paternal age and congenital malformations. Human Reprod. 2005;20(11):3173-7.
4. Decoq P, Vinckier F. Le syndrome de Down: 1. aspects médicaux. Rev Bélg Méd Dent. 1995;3:43-53.
5. Lyle R, Gehrig C, Neergaard-Henrichsen C, Deutsch S, Antonarakis SE. Gene expression from the aneuploid chromosome in a trisomy mouse model of Down syndrome. Genome Res. 2004;14(7):1268-74.
6. Mao R, WangX, Spitznagel EL Jr, Frelin LP, Ting JC, Ding H, et all. Primary and secondary transcriptional effects in the developing human Down syndrome brain and heart. Genome Biol. 2005;6(13):R107.
7. Aït Yahya-Graison E, Aubert J, Dauphinot L, Rivals I, Prieur M, Golfier G, et al. Classification of human chromosome 21 gene-expression variations in Down syndrome: impact of disease phenotypes. Am J Hum Genet. 2007;81(3):475-91.
8. Clementi M, Calzolari E, Turolla L, Volpato S, Tenconi R. Neonatal growth patterns in a population of consecutively born Down syndrome children. Am J Med Genet Suppl. 1990;7:71-4.
9. Toledo C, Alembik Y, Aguirre Jaime A, Stoll C. Growth curves of children with Down syndrome. Ann Genet. 1999;42(2):81-90.
10. Santos JA, Franceschinini SCC, Prire SE. Curvas de crescimento para crianças com síndrome de Down. Rev Bras Nutr Clin. 2006;21:144-8.
11. Cremers MJ, van der Tweel I, Boersma B, Wit JM, Zonderland M. Growth curves of Dutch children with Down's syndrome. J Intellect Disabil Res. 1996;40(Pt 5):412-20.
12. Mustacchi Z. Curvas padrão pôndero-estatural de portadores de síndrome de Down procedentes da região urbana da cidade de São Paulo [Tese de Doutorado]. São Paulo: Faculdade de Ciências Farmacêuticas da Universidade de São Paulo; 2002.
13. CDC (Centers for Disease Control and Prevention). Birth Defects. [on line] Available from: (2009 Feb 27).
14. Schuurmans FM, Pulles-Heintzberger CF, Gerver WJ, Kester AD, Forget PP. Long-term growth of children with congenital heart disease: a retrospective study. Acta Paediatr. 1998;87(12):1250-5.
15. Villasís-Keever MA, Pineda-Cruz RA, Halley-Castillo E, Alva-Espinosa C. Frecuencia y factores de riesgo asociados a desnutrición de niños com cardiopatía congénita. Salud Publica de Mex. 2001;43:313-23.
16. Peterson MB, Frantzen M, Antonarakis SE, Warren AC, Van Broeckhoven C, Chakravarti A, et al. Comparative study of microsatellite and cytogenetic markers for detecting the origin of nondisjoined chromosome 21 in Down syndrome. Am J Hum Genet. 1992;51(3):516-25.
17. Mustacchi Z, Rozone G. In: Genética baseada em evidências. [CD-ROM] São Paulo: CID Editora;1990.
18. Myrelid A, Gustafsson J, Ollars B, Annerén G. Growth charts for Down`s syndrome from birth to 18 years of age. Arch Dis Child. 2002;87(2):97-103.
19. Meguid NA, El-Kotoury AI, Abdel-Salam GM, El-Ruby MO, Afifi HH. Growth charts of Egyptian children with Down syndrome (0-36 months). East Mediterr Health J. 2004;10(1-2):106-15.
20. Cronk CE. Growth of children with Down's syndrome: birth to age 3 years. Pediatrics. 1978;61(4):564-8.
21. Freeman SB, Bean LH, Allen EG, Tinker SW, Locke AE, Druschel C, et all. Ethnicity, sex, and the incidence of congenital heart defects: a report from the National Down Syndrome Project. Genet Med. 2008;10(3):173-80.
22. Cronk C, Crocker AC, Pueschel SM, Shea AM, Zackai E, Pickens G, e al. Growth charts for children with Down syndrome: 1 month to 18 years of age. Pediatrics. 1988;81(1):102-10.
23. Dimiti AI, Anabwani GM. Anthropometric measurements in children with congenital heart disease at Kenyatta National Hospital(1985-1986). East Afr Med J. 1991;68(10):757-64.
24. Vaidynathan B, Nair SB, Sundaram KR, Babu UK, Shivaprakasha K, Rao SG, et al. Malnutrition in children with congenital heart disease (CHD) determinants and short term impact of corrective intervention. Indian Pediatr. 2008;45(7):541-6.


 All scientific articles published at rbccv.org.br are licensed under a Creative Commons license
All scientific articles published at rbccv.org.br are licensed under a Creative Commons license



 Read in English
Read in English
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket









