ABSTRACT
The events of ischemia-reperfusion injury triggers a systemic inflammatory response and can lead to cellular injury and organ failure. Such effects are noted in the post-operative recovery, especially with the use of cardiopulmonary bypass. Nowadays, it is known that leukocytes play an important role in this process. Therefore, this study addresses the role of leukocytes in the physiopathology of ischemia-reperfusion injuries and activation of inflammatory cascades through this process and seek to help in the understanding of these mechanisms as well as to bring contributions on the therapeutic approaches that can mitigate them. This retrospective review was performed from indexed scientific papers published over the last ten years in Portuguese and English in international databases MEDLINE and SciELO and related classic texts. The descriptors investigated were: ischemia-reperfusion, leukocytes, inflammatory response, cardiopulmonary bypass, adverse effects and apoptosis.
RESUMO
Os eventos de isquemia-reperfusão desencadeiam uma resposta inflamatória sistêmica que pode levar a lesões celulares e até falência de órgãos. Tais repercussões são notadas no pós-operatório de cirurgias, em especial, com o uso de circulação extracorpórea. Sabe-se, atualmente, que os leucócitos exercem importante papel neste processo. Assim, este estudo aborda o papel dos leucócitos na fisiopatologia das lesões de isquemia-reperfusão e a ativação das cascatas inflamatórias por esse processo e procura auxiliar na compreensão destes mecanismos assim como trazer contribuições acerca das abordagens terapêuticas que possam atenuá-los. Esta revisão bibliográfica retrospectiva foi realizada a partir de documentos científicos publicados nos últimos dez anos, em português e inglês, indexados em bases de dados internacionais Medline e SciELO e de textos clássicos relacionados. Os descritores pesquisados foram: isquemia-reperfusão, leucócitos, resposta inflamatória, circulação extracorpórea, efeitos adversos e apoptose.
INTRODUCTION
Cardiovascular diseases are currently the leading cause of death in the Western world. In 2005, they accounted for 30% of the total, which meant 17.5 million deaths, and 1/3 of these were caused by ischemic heart disease [1,2]. It is estimated that by the year 2015, the number of deaths will rise to 20 million, demonstrating the importance of measures that can contribute to their lower incidence and better therapy [1].
In ischemic heart disease in both acute and chronic phase, there is the potential risk for two major cases that ultimately can lead to systemic inflammatory response: 1) ischemia followed by reperfusion after cardiac obstruction of the coronary arteries and 2 ) complications arising from the use of cardiopulmonary bypass (CPB) for surgery with aortic clamping.
During ischemia, anaerobic metabolism prevails, with the increase of lactate and inorganic phosphate and decreased pH, adenosine triphosphate (ATP) and creatine. Lack of ATP leads to failure of transmembrane pumps and changes in ion gradient of cells, with the influx of sodium and calcium to the intracellular mean and cellular edema. The increase in intracellular calcium mainly activates phospholipase A
2 and calpain and other cytoplasmic proteases, while the failure of lysosomal hydrogen pumps and the decrease of pH activate lysosomal enzymes that damage cellular organelles directly. Phospholipase A
2 degrades arachidonic acid, leading to inflammatory mediators such as leukotrienes, prostaglandins and thromboxanes. The action of these substances triggers adhesion and neutrophil activation, vasoconstriction, tissue injury, platelet aggregation and chemotaxis in the ischemic area [3,4]. The calpain during ischemia transforms xanthine dehydrogenase generated by anaerobic metabolism in xanthine oxidase, which is important in reperfusion injury, as discussed below [3].
Furthermore, restoration of blood flow (reperfusion), necessary for the recovery of cellular function, may worsen the lesions present in ischemia, causing irreversible damage and cellular demise. The reintroduction of molecular oxygen (O
2) in ischemic tissue produces oxygen free radicals (OFR), highly damaging to cells that can initiate an exacerbated systemic inflammatory reaction. Specifically in cardiac surgery with cardiopulmonary bypass (CPB), cardiopulmonary ischemia inherent to surgery causes additional detrimental mechanism of ischemia and reperfusion, in both the lungs as in the heart and it causes an immune response through blood contact with synthetic materials which consists of CPB [5-7].
REPERFUSION AND LEUKOCYTES OFR
Under physiological conditions, nicotinamide adenine dinucleotide phosphate (NADPH) is the final electron acceptor in the respiratory chain, which has the participation of xanthine dehydrogenase enzyme catalyzing this reaction, whose final product is water. In the post-ischemic tissues (reperfusion), there is the accumulation of xanthine oxidase that, instead of NADPH, uses O
2, provided by reperfusion, as the final electron acceptor. In the hypoxanthine-xanthine reaction, electrons are transferred to O
2, generating the superoxide radical (O
2 -), which spontaneously or by enzymatic action (reaction with superoxide dismutase-SOD), undergoes dismutation to hydrogen peroxide (H
2O
2) (reaction 1). Through the reaction described by Haber-Weiss (2), the reaction between H
2O
2 and O
2- may give rise
in vivo to hydroxyl radical (OH
). In the presence of certain transition metals (Fe
2+, Cu
1+, Co
2+), the OH
can be formed more rapidly, by reaction 3, described by Fenton [5,8,9].
Other known routes of production of OFR are autoxidation of catecholamines and the enzyme NADPHoxidase neutrophil. Activated neutrophils generate OFR in the phagocytic vacuoles, by activation of nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase), which converts O
2 to superoxide anion (O
ÿ), followed by the chain of events until the formation of radical OH
ÿ.
Myeloperoxidase present in leukocytes catalyzes the reaction of H
2O
2 with chlorine to form sodium hypochlorite, a powerful oxidizing agent [10-12] (Figure 1).
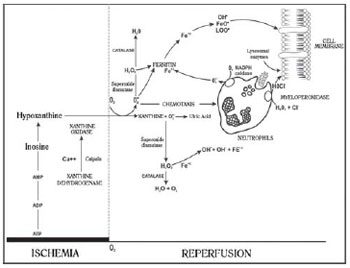 Fig. 1 - Biochemical events of ischemia-reperfusion, neutrophil activation and injury mechanisms
Fig. 1 - Biochemical events of ischemia-reperfusion, neutrophil activation and injury mechanisms
The OFR and products of inflammatory reaction function as chemotactic, attracting and activating leukocytes, which release several proteolytic enzymes such as elastases, hydrolases, myeloperoxidase and proteases, causing tissue damage and amplifying the inflammatory response and chemotaxis [3,4,6] .
CELLULAR INTERACTIONS INVOLVING THE INFLAMMATORY RESPONSE
The leukocytes are the major blood cells involved in inflammatory response, although platelets and red blood cells also participate. Leukocytes are classified as neutrophils (40% -75%), lymphocytes (20% -50%), monocytes (2% -10%), eosinophils (1% -6%) and basophils (<1%). Of these, the neutrophils are the most important in the pathogenesis of inflammation. They are the predominant cells in the first 6 to 24 hours in acute inflammation [13]. They measure from 12 to 18 micrometers, and can last for 7-10 hours in the circulation and undergo apoptosis within 24 hours.
The main changes in acute inflammation occur: 1) in the vascular caliber with vasodilatation (with or without previous transient vasoconstriction) that lead to an increase in blood flow, 2) structural changes in microcirculation which allow extravasation of plasma proteins to the interstitium in the form of inflammatory exudate (edema) and 3) migration of leukocytes in the microcirculation and their accumulation at the site of initial injury [l3-16].
Soon after this initial injury, vasodilation and increased endothelial permeability occur causing increased hydrostatic pressure and decreased plasma osmotic pressure by the outflow of fluid rich in proteins. The fluid loss results in a high concentration of red cells and increase blood viscosity, leaving the flow slower (stasis), and contributing to the leukocytes (especially neutrophils) to move to the most peripheral layers of the bloodstream, initiating the so called leukocyte margination along the vascular endothelium [6].
Due to inflammation, after the process of marginalization, both endothelial cells and circulating leukocytes are activated by circulating inflammatory substances. It starts, then, the phase of rolling leukocytes that through the exposure of its receptors, L-selectins, and their interaction with receptors P-selectin from activated endothelial cells develop a phase of loose adhesion to the endothelial cells that produce this rolling procedure [3-6]. The firm adhesion occurs later through the contact of leukocyte integrins with endothelial immunoglobulin [3-6].
Hence, at the beginning of the process there are the selectins, among them the P-selectin, which participates only in the rolling, while the E-selectin participates in both rolling and adhesion of leukocytes to the endothelium, as highlighted below:
1. P-selectin is an intracellular glycoprotein present in the platelets and endothelium, which when expressed; it binds primarily to the neutrophils, and this linkage normally is induced by the TNF and IL-1;
2. E-selectin (ELAM-1), also exposed by the activated endothelium, is responsible for the adhesion of most leukocyte groups, especially in the initial stage [6];
3. L-selectin is present on the surface of the majority of neutrophils, monocytes and lymphocytes, and it participates in the initiation of the adhesion of the leukocytes to the endothelium [6.14].
Then the process moves to the strong adhesion when integrins in leukocytes and immunoglobulins in the endothelial cells engage. Integrins are transmembrane cell surface proteins that react to signals for cell activation and bind to the immunoglobulins and the extracellular matrix.
Each integrin contains α and β chains, with characteristic structures and classified according to their β chain:
1. The β1-integrins, also called
very late antigen (VLA), are present in leukocytes and contain many subunits of α1 to α6. Integrin α4β1 (VLA-4 or CD49d) is important in relation to leukocyte-endothelial adhesion, for it interacts with the vascular cell adhesion molecule (VCAM-1);
2. The β2-integrins are rapidly presented by leukocytes in response to acute symptoms. B2-integrins are the LFA-1 (also called CD11a / CD18 or αLβ2) and MAC-1 (CD11b/ CD18 or αMβ2). The integrin MAC-1 and LFA-1 are the most studied, and the integrin MAC-1, in particular, plays an important role both in adhesion and diapedesis of all leukocytes [15,16].
The immunoglobulins are expressed on endothelial cells as receptors for leukocyte integrins. The VCAM-1 and the intracellular adhesion molecule-1 (ICAM-1) are the main receptors for the leukocyte β2-integrins [6,14,15].
After a strong adhesion to the endothelium, the leukocytes migrate through interendothelial junctions (diapedesis) and are directed to sites of inflammation driven by chemotactic factors [6] (Figure 2).
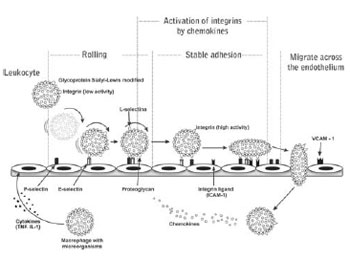 Fig. 2 - Stages of the process of leukocyte migration
Fig. 2 - Stages of the process of leukocyte migration
Both the adhesion and the leukocyte diapedesis are affected by chemical mediators of inflammation, which, besides the chemotactic effect, may generate a cascade able to expand and release other stimulating factors. The chemotaxis involves the binding of chemical mediators or chemotactic agents to specific receptors on the surface of the G protein of leukocytes that activates the inlet of the phosphatidylinositol-3 kinase (PI-3K). These changes cause increased cytosolic calcium and activate guanosine triphosphate (GTPase), favoring the production of pseudopodia and leukocyte movement. Besides locomotion, chemotactic agents also induce the activation of leukocytes with all their consequences: the production of arachidonic acid metabolites, degranulation and secretion of lysosomal enzymes, secretion of cytokines, as well as increased expression of adhesion molecules and increased exposure of integrins.
Among the mediators of inflammation there are: vasoactive amines (histamine, serotonin), arachidonic acid metabolites (prostaglandins, leukotrienes and lipoxins), plasma proteins (complement system, kinin and coagulation), platelet activating factor (PAF), cytokines (TNF and IL-1), nitric oxide (NO), leukocyte lysosomal components and OFR. Of these, the products of arachidonic acid metabolism, complement fragments and cytokines seem to have fundamental action in chemotaxis [3-5].
Histamine and serotonin are among the first chemical mediators released during inflammation. They are found in mast cells, basophils and platelets in the blood. The release of mast cells is triggered by various factors such as immunological reactions involving IgE, complement fragments C3a and C5a, cytokines (IL-1, IL-18) and histamine releasing factors derived from leukocytes. The release of platelets is stimulated after contact with collagen, thrombin, adenosine diphosphate (ADP), antigen-antibody complexes and platelet activating factors.
The metabolites of the arachidonic acid are important chemical mediators of inflammation and chemotactic agents. From arachidonic acid, through the mean of lipo-oxigenase, leukotrienes are formed, among them, leukotriene B4 (LTB4), which binds to specific receptors on the surface of leukocytes resulting in a series of responses that include activation of molecular adhesion of β
2 integrins and bond to the endothelial cell. Leukotrienes C4 and D4 and TxA
2 affect blood flow and perfusion by direct action in the microcirculation. By the cyclo-oxygenase mean are released prostacyclin (PGI
2) and thromboxane A
2 (TxA
2). The PGI
2 causes vasodilation and inhibits platelet aggregation, while the TxA
2 synthesized at the level of platelets, it causes vasoconstriction and induces strong platelet aggregation. The TxA
2 is a strong chemotactic for neutrophils and promotes the activation and adherence thereof to the endothelium [5,6].
The complement fragments are also among the most potent chemotactic agents. They are plasma proteins that when activated they become proteases that degrade other complement proteins, forming a cascade.
Complement activation may occur through several channels, including the classical (complex Ag-Acdependent, via deposition of IgM in ischemic tissues) and the alternative conduit (hydrolysis dependent), both of which cause the cleavage of C3, whose products lead to cell lysis, chemotaxis, opsonization and changes in vascular permeability. The C3a and C5a fragments are important initiators of neutrophil activation and production of interleukin-8 (IL-8) [4.17].
The pro-inflammatory cytokines are produced mainly by lymphocytes and macrophages but also by endothelial cells [18]. The two major cytokines in acute inflammation are TNF-α and interleukins (IL-1, IL-6 and IL-8), which are important endogenous mediators of adhesion molecules. Under the influence of cytokines, there is exposure of negative charges on the surface of endothelial cells (rolling and adhesion phase), activating prekallikrein, which then becomes active kallikrein and activates the factor XII, which, when activated, it activates neutrophils. Degranulation of neutrophils induced by TNF-alpha or factor XIIa, leads to the destruction of the architecture of the vascular endothelium through the release of proteolytic enzymes such as elastase and collagenase [4-6].
Most chemotactic agents have short half-life, being inactivated by enzymes or inhibitors.
Lysosomal proteins of neutrophils and monocytes, when released, can also contribute to the inflammatory response. Neutrophils have specific granules, which are smaller and contain lysozyme, collagenase, gelatinase, lactoferrin, plasminogen activator, histaminase, and larger azurophilic granules with myeloperoxidase, bactericidal factors (lysozyme, defensins), acid hydrolases and several neutral proteases (elastase, cathepsin G, collagenases). These granules may discharge their contents in phagocytic vacuoles or, alternatively, the content can be excreted directly into the extracellular space or released after cellular demise. The acid proteases degrade proteins, bacteria, and fragments only within phagolysosomes, while the neutral proteases are capable of degrading the extracellular components in a neutral pH. Lysosomal components may further increase vascular permeability and chemotaxis and also cause tissue damage [6].
Another mediator of the inflammatory response that also acts by changing tone and vascular permeability in addition to being a chemotactic agent is the nitric oxide (NO). NO is synthesized by nitric oxide synthase enzyme (NOS), present in the endothelium, which is activated by increased intracellular calcium or by macrophages after induction by certain cytokines such as interferon-γ. Besides vasodilation, inhibiting platelet aggregation and adhesion, the nitric oxide also acts as a compensatory mechanism to reduce leukocyte recruitment. NO, on the other hand, can combine with the OFR leading to the formation of metabolites such as peroxynitrite (OONO-), S-nitrosothiol, and nitrogen dioxide (NO
2 ÿ), which are antimicrobial, but run the risk of damaging host cells. The OONO
- or its decomposition products can initiate lipid peroxidation without the need for iron. There is evidence to indicate that during ischemia and reperfusion there is a close relationship between NO and endothelin [5,6,19]. Endothelin is a potent vasoconstrictor and responds to various stimuli that increase the levels of mRNA of pre-pro-peptide endothelin- 1 isoform (PPET-1) in endothelial cells, including: ischemia, thrombin, vascular injury and low levels of NO. This increase in endothelin occurs possibly from matrix metalloproteinases (gelatinase 1 and 2) that via G protein receptor of the neutrophil cell membrane leads to exposure of their â-integrins and the adhesion of neutrophils to the endothelium [20-22] (Figure 3 ).
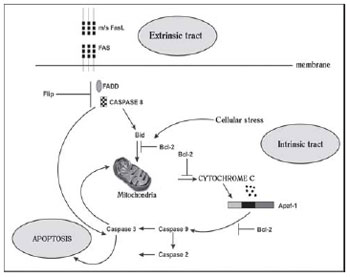 Fig. 3 - Intrinsic and extrinsic conduits of caspase activation
Fig. 3 - Intrinsic and extrinsic conduits of caspase activation
The OFR, in addition to chemical mediators of inflammatory response and activate different means that mediate both gene expression (cellular adaptation) as well as apoptosis. Low or moderate levels of OFR are controlled by endogenous antioxidant mechanisms, while high levels can damage the DNA in cells, as well as proteins and lipids, leading to responses such as proliferation, growth arrest, senescence, and even cellular demise (apoptosis) [23].
APOPTOSIS
Apoptosis is a mechanism of programmed cell death, regulated by intracellular controls that can eliminate unwanted cells without damaging neighboring cells and without causing adjacent inflammation. This is a very important physiological cell process for tissue homeostasis, but it can also be triggered by pathological conditions [6.23].
Described in 1972, it has its mechanism regulated by genetic control [24]. In this process the cellular plasma membrane remains intact while the organelles are grouped and the cell size decreases. Chromatin is linked to the nuclear membrane and fragmentation of the nucleus may occur, while blisters of cytoplasm are formed and coated by plasma membrane. These blisters break off and form apoptotic bodies, which are then recognized and phagocytosed without loss of cell membrane integrity, leakage of enzymes and local inflammatory process, which gives it significant difference between apoptosis and necrosis [6].
The effector mean of apoptosis, the caspases mean, is initiated in the cell cytoplasm through the activation of proteins of the group of cysteine proteases, called caspases (cysteine-aspartic-acid-proteases). The caspases identified in humans are eleven and divided into two types: starters such as caspases 8 and 9, and effectors, as caspase 3 and 7 [24-26]. After the activation of a caspase activator occurs, the enzyme demise program is initiated. Since the caspases will act cleaving cytoskeletal proteins and leading to the destruction of the cell nucleus. There are two caspase initiating means: the extrinsic (receptor-initiated demise) and intrinsic mean (mitochondrial). Both means converge on the effector mean [27] (Figure 3).
The internal activator of the intrinsic mean, the cytochrome c (Figure 3), responds to mitochondrial alterations, changes in electric transportation, loss of transmembrane potential, oxide reduction injury, and also the participation of the pro-and anti-apoptotic Bcl-2) protein family or DNA damage resulting from cellular toxicity or radiation. The activators of the extrinsic mean are the TNF family and bind to the receptor TNF-R1 and Fas mean. TNFR1 initiates the apoptotic process by binding to the TNF death receptor (TRADD) and Fas-death protein (FADD) [23]. The Fas receptor (APO-1or CD-95) binds to FasL, another component of the TNF group. The complex Fas and Fas-L results in the formation of the death inducing signalling complex (DISC), which contains the FADD and activates caspases 8 and 10 [25].
Caspases can be inhibited by the apoptosis inhibitory protein (AIP), which can be displaced by the second activator of caspase-derived mitochondria (SMAC) [26]. Caspase inhibitors are being studied in an attempt to reduce the inflammatory response in models of cardiac ischemiareperfusion in rats [28]. In this sense, for example, preliminary results show that intravenous administration of Z-Val-Ala- DL-Asp-fluorometilquetone (ZVAD-fmk) has contributed at least partially, to the attenuation of apoptosis of cardiomyocytes [28].
The OFR may activate numerous cell signaling means, and modulate, directly or indirectly, the functions of many enzymes and transcription factors through cascading effect signals. The size and duration of stress as well as the cell type involved are important factors to determine which mean will be activated [26].
Considering the overwhelming inflammatory network involved in ischemia-reperfusion injury, this situation seems to be even more complex specifically in cases of myocardial ischemia, when alongside the treatment of myocardial ischemia, and reperfusion, it is added the necessary use of CPB.
ADVANTAGES AND LIMITATIONS OF THE USE OF THE CPB
After more than a half century of the first cardiac surgery with CPB, its techniques were refined by the development of oxygenators, blister-catcher, biocompatible materials, filters and pumping systems with centrifugal vortex, among others, reducing the morbidity and mortality of the procedure [ 29.30]. Today, the use of CPB is indispensable routine in most cardiac surgery services in the world, enabling different surgical approaches with safety and efficacy. Even so, it possesses many undesirable effects inherent, leading to pulmonary, renal, neurological, hemodynamic and clotting complications that may lead to dysfunction and, occasionally, even organ failure, which justifies attempts to improve it even more [7 0.31 to 34].
In cardiac surgeries using CPB, there are damaging mechanisms triggered by ischemia and reperfusion, both in the lungs and heart, as well as the activation of the immune response triggered by the circuit. Mechanical changes of blood flow and hemodilution resulting from use of CPB may cause activation of polymorphonuclear neutrophils (PMN) and platelets, being that the main activating mechanism seems to be the contact of blood with artificial surfaces of CPB. During CPB, pulmonary ischemia, although partial, does much for the secondary lesions to the action of inflammatory mediators, for the flow through bronchial artery supplies only 20% of the necessity of lung tissue oxygenation [7].
The use of minimally invasive surgical techniques, such as cardiac surgery without CPB, appeared then as an alternative especially in elderly patients with multiple cardiac artery obstructions, and other comorbidities [35- 38]. Although many clinical reports relate well with these mini-invasive techniques, there are specific contraindications to be considered, such as cardiac dilatation, reoperation, pericarditis and need for cardiac remodeling [35-38].
Moreover, a review of the
American Heart Association Council which showed that the comparative results of the approach with and without CPB were similar between groups. However, the publication would not show the offpump surgery to be systematic and beneficial to the use of CPB. The study related as parameters; hospitalization, mortality, neurological function and cardiac performance after surgery [38].
ATTENUATION OF THE INFLAMMATORY RESPONSE: ACTIONS ON NEUTROPHILS
With the same goal of attenuating the inflammatory response and ischemia-reperfusion injuries, other approaches have been proposed, such as production of neutropenia, the action of drugs acting on leukocyte adherence and changes in cardioplegic solutions aiming cell protection.
The promotion of neutropenia by leukocyte depletion filter (LDF) [11] and radiation [39], the association between NO and hypothermia during CPB reducing the adherence of leukocytes to the endothelium [40], the modifications of the cardioplegic solution [41], heparinization of the surface of the CPB system, the addition of anti-inflammatory and chelating agents of OFR to the circuit [4.42 to 44], were not sufficient to reduce inflammatory markers in the postoperative period and to support the standardization of with any of these alternatives to the procedure of CPB [45].
In 1972, there were marketed the first FDLS [46]. In the late 90's, they were widely used in CPB during cardioplegia in both the transfused blood and blood drawn into the circuit or in preservation solutions [47-49].
Initial clinical studies showed beneficial effects of LDF on CPB, with lower release of leukocyte enzymes, improvement in clinical recovery, a shorter stay in the intensive care unit (ICU) and lower costs [50-52]. However, other authors showed that the use of FDL seemed to be less effective than expected, and its effectiveness diminished over surgical time [53-55]. They also observed that leukocytes trapped in the meshes of the filter could be activated, releasing their products into the circulation [56.57]. Hence, from that point on, the FLDs were used less due to lack of sufficient evidence about their benefit [58].
The immunoregulatory approach of apoptosis represents a new aspect to control inflammation associated with CPB. For it is known today that the membrane receptor Fas (CD95) in conjunction with Fas ligand (FasL), a protein produced by immune cells, in addition to inducing apoptosis plays an important role in regulating cell proliferation and tumor growth. Thus, a dysfunction of the Fas/FasL in tissue cells by delaying apoptosis, may promote proliferative disorders, while a lower apoptotic activity of this system in PMNs, may have some therapeutic potential antiviral or anticancer effect [18,59,60] .
By interfering in the life span of cells, the receptors Fas/ FasL can act as regulators of responses mediated by PMNs [61]. Increased survival of PMNs can cause great tissue damage by longer secretion of toxic metabolites [62], while a shorter life span can mitigate immune reactions [59,60].
The therapeutic potential of an anti-Fas antibody in rapid inactivation of neutrophils was noted in
in vitro studies,
in vivo studies in animals and clinical studies of patients undergoing cardiac surgery [63-65]. This group of researchers has developed a leukocyte inhibition module (
leukocyte inhibition module-LIM
®), apparently similar to a biological filter, but with the plot covered by anti-Fas.
This module, when inserted in the CPB, resulted in decreased neutrophil hyperactivity, with significant activation of apoptosis of theses cells [65-68].
Although this approach appears promising by reducing the amount of inflammatory markers and showing good cardiac recovery in both animal models and in clinical surgery in humans, it requires further studies with larger numbers of cases.
CONCLUSION
The great size of the secondary inflammatory response to ischemia-reperfusion and the use of CPB indicates the need for measures that might if not inhibit it at least mitigate it. Thus, the control of risk factors, the reduction of ischemic cardiovascular events, technical training for off-pump surgery, as well as advances in anti-inflammatory therapy, are measures to be reinforced while research should be encouraged so that these objectives are achieved.
Although the use of leukocyte filters and blocking drugs of inflammatory mediators have not been proven definitive in attenuating ischemia-reperfusion injuries, the use of combined techniques seems more promising to intervene at different points in the inflammatory network.
The use of leukocyte inhibition module that acts on the complex Fas/FasL by binding anti-CD95 immunoglobulin to the leukocyte CD-95 (LIM) has proven a safe therapeutic procedure, with the exclusive immobilization of activated leukocytes without interfering with the number of lymphocytes and without changing the patient's coagulation system.
The immunoregulatory approach can bring new perspectives, either by acting directly by means of antibodies against inflammatory mediators or in the modulation of cell membrane receptors responsible for activation, adhesion, diapedesis and leukocyte chemotaxis and also through the activation or inhibition of pro-and anti-apoptotic protein effector means, establishing itself as a field for continuous deepening.
REFERENCES
1. World Health Organization. Programmes and Projects: Cardiovascular disease [site na Internet]. Ginebra: WHO; 2009. [cited 2009 Apr 17]. Disponível em: http://www.who.int/cardiovascular_diseases/en/
2. Ministério da Saúde (BR). DATASUS. Indicadores de mortalidade: taxa de mortalidade específica por doenças do aparelho circulatório [site na Internet]. Brasília: DATASUS; 2009. [citado 2009 abr 17]. Disponível em: http://tabnet.datasus.gov.br/cgi/tabcgi.exe?idb2007/c08.def
3. Silveira M, Yoshida WB. Isquemia e reperfusão em músculo esquelético: mecanismo de lesão e perspectivas de tratamento. J Vasc Br. 2004;3(4):367-78.
4. Moura HV, Pomerantzeff PMA, Gomes WJ. Síndrome da resposta inflamatória sistêmica na circulação extracorpórea: papel das interleucinas. Rev Bras Cir Cardiovasc. 2001;16(4):376-87. View article
5. Yoshida WB. Radicais livres na síndrome da isquemia e reperfusão. Cir Vasc Angiol. 1996;12:82-95.
6. Mitchell RN, Kumar V,Abbas AK, Fausto N. Inflamação aguda e crônica. In: Robbins & Cotran: fundamentos de patologia. 7ª ed. Rio de Janeiro: Elsevier; 2006. p.29-54.
7. Clark SC. Lung injury after cardiopulmonary bypass. Perfusion. 2006;21(4):225-8. [MedLine]
8. Granger DN. Role of xanthine oxidase and granulocytes in ischemia-reperfusion injury. Am J Physiol. 1988;255(6 Pt 2):H1269-75.
9. Grace PA. Ischemia-reperfusion injury. Br J Surg. 1994;81(5):637-47. [MedLine]
10. Del Maestro RF, Planker M, Arfors KE. Evidence for the participation of superoxide anion radical in altering the adhesive interaction between granulocytes and endothelium, in vivo. Int J Microcirc Clin Exp. 1982;1(2):105-20. [MedLine]
11. Johnson D, Thomson D, Hurst T, Prasad K, Wilson T, Murphy F, et al. Neutrophil-mediated acute lung injury after extracorporeal perfusion. J Thorac Cardiovasc Surg. 1994;107(5):1193-202. [MedLine]
12. Kirschner RE, Fantini GA. Role of iron and oxygen-derived free radicals in ischemia-reperfusion injury. J Am Coll Surg. 1994;179(1):103-17. [MedLine]
13. Junqueira LC, Carneiro C. Histologia básica. 10ª ed. Rio de Janeiro:Guanabara Koogan;2004. p.223-37.
14. Bevilacqua MP, Nelson RM. Selectins. J Clin Invest. 1993;91(2):379-87. [MedLine]
15. Stewart M, Thiel M, Hogg N. Leukocyte integrins. Curr Opin Cell Biol. 1995;7(5):690-6. [MedLine]
16. Lu H, Ballantyne C, Smith CW. LFA-1 (CD11a/CD18) triggers hydrogen peroxide production by canine neutrophils. J Leukoc Biol. 2000;68(1):73-80. [MedLine]
17. Gourlay T, Asimakopoulos G, Taylor KM. Leukocyte biology and pathogenicity in cardiac surgery and cardiology: the need for leukocyte depletion. In: Matheis G, Moritz A, Scholz M, eds. Leukocyte depletion in cardiac surgery and cardiology. Basel:Karger;2002. p.1-12.
18. Mojcik CF, Levy JH. Aprotinin and the systemic inflammatory response after cardiopulmonary bypass. Ann Thorac Surg. 2001;71(2):745-54. [MedLine]
19. Cerqueira NF, Yoshida WB. Óxido nítrico. Revisão. Acta Cir Bras. 2002;17(6):417-42.
20. Gianetti J, Del Sarto P, Bevilacqua S, Vassalle C, De Filippis R, Kacila M, et al. Supplemental nitric oxide and its effect on myocardial injury and function in patients undergoing cardiac surgery with extracorporeal circulation. J Thorac Cardiovasc Surg. 2004;127(1):44-50. [MedLine]
21. Carvalho MHC, Nigro D, Lemos VS, Tostes RCA, Fortes ZB. Hipertensão arterial: o endotélio e suas múltiplas funções. Rev Bras Hipertens. 2001;8(1):76-88.
22. Fernandez-Patron C, Zouki C, Whittal R, Chan JS, Davidge ST, Filep JG. Matrix metalloproteinases regulate neutrophil-endothelial cell adhesion through generation of endothelin-1[1-32]. FASEB. 2001;15(12):2230-40.
23. Gulbins E, Jekle A, Ferlinz K, Grassmé H, Lang F. Physiology of apoptosis. Am J Physiol Renal Physiol. 2000;279(4):F605-15.
24. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26(4):239-57. [MedLine]
25. Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3(9):745-56. [MedLine]
26. Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002;192(1):1-15. [MedLine]
27. Mitchell RN, Kumar V, Abbas AK, Fausto N. Adaptações celulares, lesão celular e morte celular. In: Robbins & Cotran: fundamentos de patologia. 7ª ed. Rio de Janeiro: Elsevier;2006. p.3-28.
28. Yaoita H, Ogawa K, Maehara K, Maruyama Y. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation. 1998;97(3):276-81. [MedLine]
29. Stephenson LH. History of cardiac surgery. In: Cohn LH, Edmunds LH Jr., eds. Cardiac surgery in the adult. New York:McGraw-Hill;2003. p.329.
30. Gomes WJ, Saba JC, Buffolo E. 50 anos de circulação extracorpórea no Brasil: Hugo J. Felipozzi, o pioneiro da circulação extracorpórea no Brasil. Rev Bras Cir Cardiovasc. 2005;20(4):3-8. View article
31. Brasil LA, Gomes WJ, Salomão R, Buffolo E. Ativação de citocina (fator de necrose tumoral - alfa) e resposta clínica induzida pela circulação 32. extracorpórea. Rev Bras Cir Cardiovasc. 1996;11(3):188-200.
32. Sievers HH, Freund-Kaas C, Eleftheriadis S, Fischer T, Kuppe H, Kraatz EG, et al. Lung protection during total cardiopulmonary bypass by isolated lung perfusion: preliminary results of a novel perfusion strategy. Ann Thorac Surg. 2002;74(4):1167-72.
33. Wang QP, Gu JW, Zhan XH, Li H, Luo XH. Assessment of glomerular filtration rate by serum cystatin C in patients undergoing coronary artery bypass grafting. Ann Clin Biochem. 2009;46(Pt 6):495-500. [MedLine]
34. Gomes WJ, Erlichman MR, Batista-Filho ML, Knobel M, Almeida DR, Carvalho AC, et al. Vasoplegic syndrome after off-pump coronary artery bypass surgery. Eur J Cardiothorac Surg. 2003;23(2):165-9. [MedLine]
35. Buffolo E, Branco JN, Gerola LR, Aguiar LF, Teles CA, Palma JH, et al. Off-pump myocardial revascularization: critical analysis of 23 years? experience in 3,866 patients. Ann Thorac Surg. 2006;81(1):85-9. [MedLine]
36. Brasil LA, Gomes WJ, Salomão R, Buffolo E. Inflammatory response after myocardial revascularization with or without cardiopulmonary bypass. Ann Thorac Surg. 1998;66(1):56-9. [MedLine]
37. Lobo Filho JG, Leitão MCA, Lobo Filho HG, Soares JPH, Magalhães GA, Leão Filho CSC, et al. Cirurgia de revascularização coronariana esquerda sem CEC e sem manuseio da aorta em pacientes acima de 75 anos. Rev Bras Cir Cardiovasc. 2002;17(3):208-14. View article
38. Selke FW, DiMaio JM, Caplan LR, Ferguson TB, Gardner TJ, Hiratzka LF, et al. Comparing on-pump and off-pump coronary artery bypass grafting: numerous studies but few conclusions: a scientific statement from the American Heart Association council on cardiovascular surgery and anesthesia in collaboration with the interdisciplinary working group on quality of care and outcomes research. Circulation. 2005;111(21):2858-64. [MedLine]
39. Belkin M, LaMorte WL, Wright JG, Hobson RW 2nd. The role of leukocytes in the pathophysiology of skeletal muscle ischemic injury. J Vasc Surg. 1989;10(1):14-8.
40. el Habbal MH, Carter H, Smith LJ, Elliott MJ, Strobel S. Neutrophil activation in paediatric extracorporeal circuits: effect of circulation and temperature variation. Cardiovasc Res. 1995;29(1):102-7. [MedLine]
41. Dussin LH, Moura L, Gib MC, Saadi EK, Barbosa GV, Wender OCB. Análise ultra-estrutural do miocárdio usando solução cardioplégica cristalóide com e sem procaína em pacientes submetidos à troca valvar aórtica. Rev Bras Cir Cardiovasc. 2008;23(3):389-5. View article
42. Roberts I, Yates D, Sandercock P, Farrell B, Wasserberg J, Lomas G, et al; CRASH trial collaborators. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): randomised placebo-controlled trial. Lancet. 2004;364(9442):1321-8. [MedLine]
43. Havlicek K, Motycka V, Siller J, Cervinka V. Systemic inflammatory response syndrome (SIRS) in serious chest injuries: is a pharmacological blockade effective? Ann Thorac Cardiovasc Surg. 2005;11(4):232-7. [MedLine]
44. Goebel U, Siepe M, Mecklenburg A, Doenst T, Beyersdorf F, Loop T, et al. Reduced pulmonary inflammatory response during cardiopulmonary bypass: effects of combined pulmonary perfusion and carbon monoxide inhalation. Eur J Cardiothorac Surg. 2008;34(6):1165-72. [MedLine]
45. Bartels C, Gerdes A, Babin-Ebell J, Beyersdorf F, Boeken U, Doenst T, et al. Cardiopulmonary bypass: Evidence or experience based? J Cardiovasc Surg. 2002;124(1):20-7.
46. Greenwalt TJ, Gajewski M, McKenna JL. A new method for preparing buffy coat-poor blood. Transfusion. 1962;2:221-9. [MedLine]
47. Roth M, Kraus B, Scheffold T, Reuthebuch O, Klövekorn WP, Bauer EP. The effect of leukocyte-depleted blood cardioplegia in patients with severe left ventricular dysfunction: a randomized, double-blind study. J Thorac Cardiovasc Surg. 2000;120(4):642-50. [MedLine]
48. van de Watering LM, Hermans J, Houbiers JG, van den Broek PJ, Bouter H, Boer F, et al. Beneficial effects of leukocytes depletion of transfused blood on postoperative complications in patients undergoing cardiac surgery: a randomized clinical trial. Circulation. 1998;97(6):562-8. [MedLine]
49. Lick SD, Brown PS Jr, Kurusz M, Vertrees RA, McQuitty CK, Johnston WE. Technique of controlled reperfusion of the transplanted lung in humans. Ann Thorac Surg. 2000;69(3):910-2. [MedLine]
50. Alexiou C, Tang AA, Sheppard SV, Smith DC, Gibbs R, Livesey SA, et al. The effect of leucodepletion on leucocyte activation, pulmonary inflammation and respiratory index in surgery for coronary revascularisation: a prospective randomised study. Eur J Cardiothorac Surg. 2004;26(2):294-300. [MedLine]
51. Olivencia-Yurvati AH, Ferrara CA, Tierney N, Wallace N, Mallet RT. Strategic leukocyte depletion reduces pulmonary microvascular pressure and improves pulmonary status post-cardiopulmonary bypass. Perfusion. 2003;18(Suppl 1):23-31. [MedLine]
52. Kiliç D, Gunaydin S, Kisa U, Sari T, Deveci O, Zorlutuna Y. Clinical efficacy of leukofiltration on cardiopulmonary bypass related inflammatory response: Fact or Foe? Inflamm Res. 2009;58(6):292-7. [MedLine]
53. Leal-Noval SR, Amaya R, Herruzo A, Hernández A, Ordóñez A, Marín-Niebla A, et al. Effects of a leukocyte depleting arterial line filter on perioperative morbidity in patients undergoing cardiac surgery: a controlled randomized trial. Ann Thorac Surg. 2005;80(4):1394-400. [MedLine]
54. Baksaas ST, Flom-Halvorsen HI, Ovrum E, Videm V, Mollnes TE, Brosstad F, et al. Leucocyte filtration during cardiopulmonary reperfusion in coronary artery bypass surgery. Perfusion. 1999;14(2):107-17. [MedLine]
55. Smit JJ, de Vries AJ, Gu YJ, van Oeveren W. Efficiency and safety of leukocyte filtration during cardiopulmonary bypass for cardiac surgery. Transfus Sci. 1999;20(3):151-65. [MedLine]
56. Scholz M, Simon A, Matheis G, Dzemali O, Henrich D, Kleine P, et al. Leukocyte filtration fails to limit functional neutrophil activity during cardiac surgery. Inflamm Res. 2002;51(7):363-8. [MedLine]
57. Ilmakunnas M, Pesonen EJ, Ahonen J, Rämö J, Siitonen S, Repo H. Activation of neutrophils and monocytes by a leukocyte-depleting filter used throughout cardiopulmonary bypass. J Thoracic Cardiovasc Surg. 2005;129(4):851-9.
58. Warren O, Alexiou C, Massey R, Leff D, Purkayastha S, Kinross J, et al. The effects of various leukocyte filtration strategies in cardiac surgery. Eur J Cardiothorac Surg. 2007;31(4):665-76. [MedLine]
59. Los M, Burek CJ, Stroh C, Benedyk K, Hug H, Mackiewicz A. Anticancer drugs of tomorrow: apoptotic pathways as targets for drug design. Drug Discov Today. 2003;8(2):67-77. [MedLine]
60. Everett H, McFadden G.Apoptosis: an innate immune response to virus infection. Trends Microbiol. 1999;7(4):160-5. [MedLine]
61. Scheel-Toellner D, Wang K, Craddock R, Webb PR, McGettrick HM, Assi LK, et al. Reactive oxygen species limit neutrophil life span by activating death receptor signaling. Blood. 2004;104(8):2557-64. [MedLine]
62. Albelda SM, Smith CW, Ward PA. Adhesion molecules and inflammatory injury. FASEB J. 1994;8(8):504-12. [MedLine]
63. Scholz M, Simon A, Berg M, Schuller AM, Hacibayramoglu M, Margraf S, et al. In vivo inhibition of neutrophil activity by a FAS (CD95) stimulating module: arterial in-line application in a porcine cardiac surgery model. J Thorac Cardiovasc Surg. 2004;127(6):1735-42. [MedLine]
64. Lögters TT, Altrichter J, Paunel-Görgülü A, Sager M, Witte I, Ott A, et al. Extracorporeal immune therapy with immobilized agonistic anti-Fas antibodies leads to transient reduction of circulating neutrophil numbers and limits tissue damage after hemmorrhagic shock/resuscitation in a porcine model. J Inflamm (Lond). 2010;7:18.
65. Moreno JB, Margraf S, Schuller AM, Simon A, Moritz A, Scholz M. Inhibition of neutrophil activity in cardiac surgery with cardiopulmonary bypass: a novel strategy with the leukocyte inhibition module. Perfusion. 2004;19(1):11-6. [MedLine]
66. Greenstein S, Barnard J, Zhou K, Fong M, Hendey B. Fas activation reduces neutrophil adhesion to endothelial cells. J Leukoc Biol. 2000;68(5):715-22. [MedLine]
67. Curtin JF, Cotter TG. Live an let die: regulatory mechanisms in Fas-mediated apoptosis. Cell Signal. 2003;15(11):983-92. [MedLine]
68. Abdel-Rahman U, Margraf S, Aybek T, Lögters T, Bitu-Moreno J, Francischetti I, et al. Inhibition of neutrophil activity improves cardiac function after cardiopulmonary bypass. J Inflamm (Lond). 2007;4:21.
Article receive on Friday, August 27, 2010
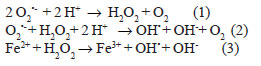
 All scientific articles published at rbccv.org.br are licensed under a Creative Commons license
All scientific articles published at rbccv.org.br are licensed under a Creative Commons license



 Read in Portuguese
Read in Portuguese
 Portuguese PDF
Portuguese PDF
 Print
Print
 Send this article by email
Send this article by email
 How to cite this article
How to cite this article
 Submit a comment
Submit a comment
 Mendeley
Mendeley
 Pocket
Pocket









